MD2B - Introduction to Oncology - Cellular and Genetic Basis of Cancer
encontrar mi
Definition
Cancer can be defined on a cellular level as a derangement in the normal regulation of cell proliferation, death and differentiation. One can view normal cell growth as a series of states, collectively termed the "cell cycle", through which a cell passes as it grows, proliferates and reaches a differentiated level. These states are identified and distinguished according to various biochemical and morphologic criteria.
Cell Cycle
The stages of the cycle can be viewed as a continuum, and thus there are not strict "starting" or "ending" points, per se, but a series of phases.
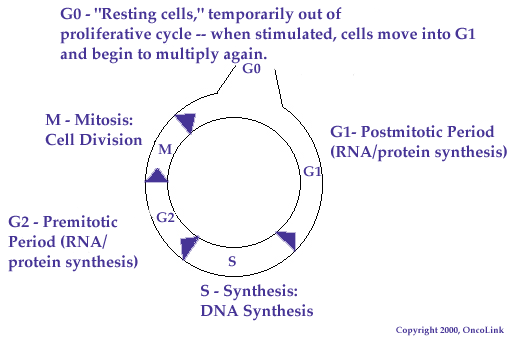
During the S phase, new DNA is synthesized and the cellular DNA content is thus duplicated in anticipation of division. Following DNA replication is phase G2, a relatively brief period of final preparation for the next phase, mitosis (M). The M phase features four substages, prophase, metaphase, anaphase and telophase. These conclude with cytokinesis and result in two progeny cells for every one mother cell. The cell next enters either G0, which is a "resting phase" of variable duration during which the cell withdraws from proliferation via the cell cycle but maintains other basic functions, or G1, a "growth" phase also of variable duration but in which the cell can either exit into G0 remain within the cycle and prepare for DNA replication, depending on the input of regulatory factors.
Important regulation points that determine the rate of cellular proliferation occur in G1, before S, and between G0 and G1. These control points serve as the focus of much cancer research, as tumor growth is thought to result from the dysregulations that can occur here.
Cellular Proto-Oncogenes
Various genes result in products responsible for overseeing cell cycle activity. Some of these products can be described as "pro-proliferation", as they enhance the rate of cell cycling or increase the rate of cellular entry into the cycle. In contrast, some gene products are considered "anti-proliferation", as they promote decreased cellular division and proliferation. Either excessive activity of the first group and/or decreased or absent activity of the second would therefore make cells behave "cancer-like" and contribute to a neoplastic phenotype.
The term "proto-oncogene" refers to a normal cellular gene that has the potential to cause neoplastic transformation when they are abnormally altered or mutated. Once "activated", ("gain of function") the gene is labeled an "oncogene". When an oncogene contributes to cellular neoplastic transformation even in the presence of normal copies of itself, it is considered to be genetically dominant, and thus called a "dominant oncogene".
Alternatively, cellular genes whose activities normally prevent neoplastic behavior are called "tumor suppressor genes". Rather than through "activation", it is their inactivity or absence ("loss of function") that these can contribute to oncogenic transformation, and hence they are called "recessive oncogenes".
Dominant Oncogenes
The activity of "dominant oncogenes" has been known for decades, as they are readily detected in vitro. Up to 60 different members have been identified. However, the relevance of these genes to human cancers has only been defined for a small subset, notably ras, myc, bcl2, and bcr-abl. It is important to remember abnormal cellular accumulation can result not only from uncontrolled cellular proliferation, but also from abnormal cell removal. Hence, dominant oncogenes often encode for apoptosis-regulating proteins (proteins that help to regulate programmed cellular death) as well as extracellular growth and intranuclear transcription factors. Some examples of activation mechanisms of dominant oncogenes are:
Chromosomal Translocations | ||
Disease Phenotype | Translocation | Mechanism |
Burkitt's lymphoma | t(8;14) (myc/Ig) | deregulated expression |
Nodular B-cell lymphomas | t(14;18) (Ig/bcl2) | deregulated expression |
Chronic myelogenous leukemia | t(9;22) (abl/bcr) | altered gene product |
Gene amplification | ||
deregulated expression | ||
Neuroblastoma (N-myc) | deregulated expression | |
Mammary carcinomas (neu/HER2) | deregulated expression | |
Point mutation | ||
Various tumors(ras) | altered gene product | |
Proviral insertion | ||
Many animal models | deregulated expression |
Tumor Suppressor Genes
Evidence for the presence of "tumor suppressor genes" has only recently emerged, with relatively few examples identified to date. This is mainly due to the inherent difficulty in detecting their activity in vitro, since detection requires loss of both copies of the gene (hence they are called "recessive oncogenes"). A single good copy of a tumor suppressor gene would usually be sufficient to suppress tumorigenesis. Some of these genes (p53 in particular) represent those most commonly mutated in up to 50% of human cancers, and many familial cancer predisposition syndromes and "inherited cancers" are clearly linked to germline loss of tumor suppessor genes, (the "double hit" hypothesis). Examples include:
Inherited predisposition | Tumor suppressor gene |
Retinoblastoma (also osteosarcoma) | Rb |
Li-Fraumeni syndrome (various carcinomas) | p53 |
Breast cancer (also ovarian) | BRCA1, BRCA2 |
Melanoma | p16INK4 |
Familial adenomatous polyposis (FAP) syndrome | APC |
Hereditary nonpolyposis colon cancer (HNPCC) | MSH1 |
Many of these genes normally prevent cells from entering the cell cycle by binding and thus inactivating transcription factors needed for cell cycling. The Rb protein is a prime example. Other tumor suppressors do not block transcription directly, but are functionally linked to these blockers and thus act indirectly. An example of this would be kinase inhibitors like p16INK4, which prevent phosphorylation and inactivation of the blocking proteins described above. Another example is the p53 gene product, which leads to selective apoptosis (programmed cell-death) as well as arrest of cell proliferation when induced by DNA damaging agents such as radiation and certain drugs. Thus, loss of p53 activity means defective apoptosis and loss of cell cycle arrest following DNA damage, minimizing time for DNA repair and promoting genomic instability.
Clinical Relevance and Future Directions
Advancements in our understanding and elucidation of the genetic and molecular pathogenesis of cancer are crucial to the advancement of clinical oncology. Research in this area is the key to improved detection, diagnosis and management of many cancers. Potential present and future applications of the knowledge such research provides include the following:
Molecular diagnostics
- Cancer classification (eg. bcr-abl in CML, Ig-myc in Burkitt's lymphoma, etc.)
- Early detection (eg. FAP, HNPCC)
- Detection of minimal residual disease
- Risk analysis in familial cancer predisposition syndromes (eg. BRCA1)
Prognostic indicators
- Ki-ras mutations in lung adenocarcinoma (present in 40%, associated with poorer prognosis)
- neu/HER2 amplification in breast cancer (present in 30%, associated with poorer prognosis)
- N-myc amplification in neuroblastoma (advanced stage of disease)
Therapeutic response indicators
- p53 mutations (eg. breast cancer)
Targets of anti-tumor therapy
- Oncogene or mRNA targeting (eg. antisense oligonucleotide therapy)
- Oncoprotein targeting
- Pharmacologic (eg. farnesyltransferase G-protein inhibitors)
- Immunologic (eg. mutant ras or p53 mutations)