Instituto Nacional del Cáncer
encontrar mi
Fecha de publicación: Jul 4, 2024
Información sobre el tratamiento de la leucemia mielomonocítica juvenile (LMMJ o JMML): trasplante de células madre y cuidados médicos de apoyo. Resumen para profesionales de la salud.
Tratamiento de la leucemia mielomonocítica juvenil
Incidencia
La leucemia mielomonocítica juvenil (LMMJ) es una leucemia rara, alrededor de 10 veces menos frecuente que la leucemia mieloide aguda en niños. La incidencia anual es de 1 a 2 casos por 1 millón de personas. La LMMJ es la neoplasia mieloproliferativa más común en los niños pequeños y se presenta a una mediana de edad de aproximadamente 1,8 años. Es más frecuente en varones (proporción entre hombres y mujeres, cerca de 2,5:1).
References
- Passmore SJ, Chessells JM, Kempski H, et al.: Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia in the UK: a population-based study of incidence and survival. Br J Haematol 121 (5): 758-67, 2003.
Cuadro clínico inicial
Las manifestaciones clínicas comunes en el momento del diagnóstico son las siguientes:
- Hepatoesplenomegalia (97 %).
- Linfadenopatía (76 %).
- Palidez (64 %).
- Fiebre (54 %).
- Erupción cutánea (36 %).
En ocasiones, los pacientes también presentan un recuento elevado de glóbulos blancos y un aumento de los monocitos circulantes.
References
- Niemeyer CM, Arico M, Basso G, et al.: Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89 (10): 3534-43, 1997.
Clasificación de la Organización Mundial de la Salud
La Organización Mundial de la Salud (OMS) clasifica la leucemia mielomonocítica juvenil (LMMJ) como una neoplasia mieloproliferativa (NMP) impulsada por la activación de la vía RAS en la primera infancia.
Para obtener información sobre el sistema de clasificación de la leucemia mieloide aguda (LMA), consultar la sección Sistema de clasificación de la Organización Mundial de la Salud para la leucemia mieloide aguda infantil en Tratamiento de la leucemia mieloide aguda infantil.
References
- Khoury JD, Solary E, Abla O, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 36 (7): 1703-1719, 2022.
Criterios diagnósticos
En el Cuadro 1 se describen los criterios que se utilizan en la actualidad para el diagnóstico definitivo de niños que presentan las manifestaciones clínicas indicativas de leucemia mielomonocítica juvenil (LMMJ).
| GM-CSF = factor estimulante de colonias de granulocitos y macrófagos; LMMJ = leucemia mielomonocítica juvenil; OMS = Organización Mundial de la Salud. | ||
| aLas variantes germinales de PTPN11, KRAS o NRAS (que causan el síndrome de Noonan) pueden producir un trastorno mieloproliferativo transitorio similar a la LMMJ. | ||
| bHay casos aislados con variantes heterocigotas en el sitio de empalme. | ||
| cComo RRAS o RRAS2. | ||
| dLos casos que no cumplen los criterios genéticos o para los que no se dispone de pruebas genéticas, deben cumplir con lo siguiente además de los criterios clínicos, hematológicos y de laboratorio. | ||
| Criterios clínicos, hematológicos y de laboratorio (todos los criterios son necesarios para el diagnóstico) | ||
| 1. Recuento de monocitos en sangre periférica ≥1 × 109/l | ||
| 2. Los blastocitos y los promonocitos constituyen <20 % de la sangre periférica y la médula ósea | ||
| 3. Evidencia clínica de infiltración de órganos, con mayor frecuencia esplenomegalia | ||
| 4. Ausencia del gen de fusión BCR::ABL1 | ||
| 5. Ausencia de reordenamiento de KMT2A | ||
| Criterios genéticos (un criterio es suficiente para el diagnóstico) | ||
| 1. Una variante de un componente o un regulador de la vía RAS canónica: | ||
| a) Una variante somática clonal de PTPN11, KRAS o NRASa | ||
| b) Una variante somática clonal o germinal de NF1 y pérdida de heterocigosidad o heterocigosis compuesta en NF1 | ||
| c) Una variante somática clonal o germinal de CBL y pérdida de heterocigosidad en CBLb | ||
| 2. Una variante patogénica clonal de la vía RAS no canónicac o fusiones que activan genes ubicados secuencia arriba en la vía RAS, como ALK, PDGFRB y ROS1 | ||
| Otros criterios (se requieren dos o más para el diagnóstico)d | ||
| 1. Precursores mieloides (promielocitos, mielocitos, metamielocitos) y eritroides circulantes | ||
| 2. Hemoglobina F elevada para la edad | ||
| 3. Trombocitopenia con médula ósea hipercelular, a menudo con hipoplasia megacariocítica. Las características displásicas no siempre son evidentes | ||
| 4. Los progenitores mieloides son hipersensibles al GM-CSF (se detectan mediante ensayos clonogénicos o al medir la fosforilación STAT5 en ausencia de GM-CSF exógeno o con dosis bajas de este) | ||
References
- Khoury JD, Solary E, Abla O, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 36 (7): 1703-1719, 2022.
Patogénesis y factores de riesgo
La patogénesis de la leucemia mielomonocítica juvenil (LMMJ) se ha relacionado de forma estrecha con la activación de la vía del oncogén RAS, así como con síndromes relacionados (consultar la Figura 1). Además, se notificaron patrones característicos de expresión del RNA y metilación del DNA. Estos patrones se correlacionan con factores clínicos como la edad, que repercuten sobre el pronóstico.
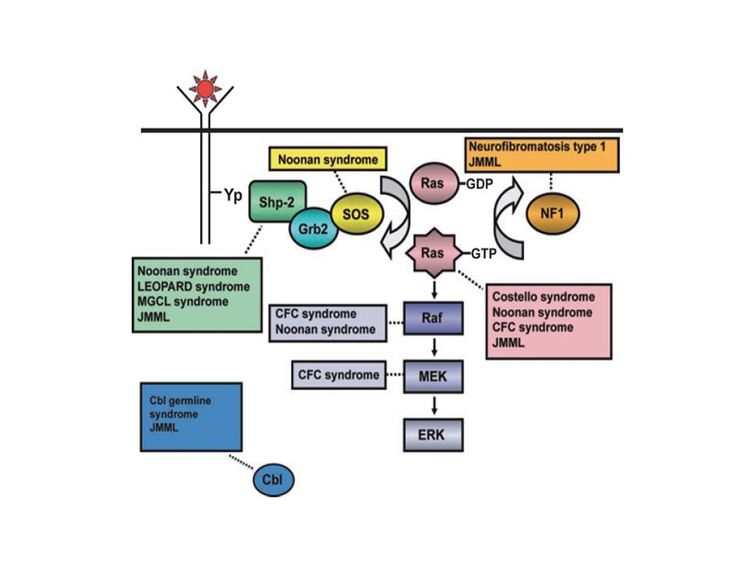 Figura 1. Diagrama esquemático que muestra la activación de Ras estimulada por un ligando, la vía Ras-Erk y las mutaciones génicas identificadas hasta la fecha que contribuyen con el trastorno congénito neuro-cardio-facio-cutáneo y la LMMJ. NL/MGCL: lesiones múltiples de células gigantes tipo Noonan; CFC: cardio-facio-cutáneo; LMMJ: leucemia mielomonocítica juvenil. Reproducido de Leukemia Research, 33 (3), Rebecca J. Chan, Todd Cooper, Christian P. Kratz, Brian Weiss, Mignon L. Loh, Juvenile myelomonocytic leukemia: A report from the 2nd International JMML Symposium, Pages 355-62, Derechos de autor 2009, con permiso de Elsevier.
Figura 1. Diagrama esquemático que muestra la activación de Ras estimulada por un ligando, la vía Ras-Erk y las mutaciones génicas identificadas hasta la fecha que contribuyen con el trastorno congénito neuro-cardio-facio-cutáneo y la LMMJ. NL/MGCL: lesiones múltiples de células gigantes tipo Noonan; CFC: cardio-facio-cutáneo; LMMJ: leucemia mielomonocítica juvenil. Reproducido de Leukemia Research, 33 (3), Rebecca J. Chan, Todd Cooper, Christian P. Kratz, Brian Weiss, Mignon L. Loh, Juvenile myelomonocytic leukemia: A report from the 2nd International JMML Symposium, Pages 355-62, Derechos de autor 2009, con permiso de Elsevier.
Los síndromes y las características genéticas vinculadas con un aumento del riesgo de LMMJ son los siguientes:
- Neurofibromatosis de tipo 1 (NF1). Hasta un 14 % de los casos de LMMJ se presentan en niños con NF1.
- Síndrome de Noonan. El síndrome de Noonan a menudo se hereda como una enfermedad autosómica dominante, pero también surge de manera espontánea. Se caracteriza por dismorfia facial, estatura baja, cuello alado, así como anomalías neurocognitivas y cardíacas. Se observan variantes germinales de PTPN11 en niños con síndrome de Noonan y en niños con LMMJ.
Es importante destacar que algunos niños con síndrome de Noonan tienen características hematológicas indistinguibles de la LMMJ que remiten de forma espontánea durante la lactancia, parecido a lo que ocurre en los niños con síndrome de Down y trastorno mieloproliferativo transitorio.
En una cohorte prospectiva numerosa de 641 pacientes con síndrome de Noonan y una variante germinal de PTPN11, se encontró que 36 pacientes (cerca del 6 %) presentaban características mieloproliferativas; 20 pacientes (cerca del 3 %) cumplieron con los criterios diagnósticos de consenso para la LMMJ.
- De los 20 pacientes que cumplían con los criterios de LMMJ, 12 pacientes tenían manifestaciones neonatales graves (por ejemplo, complicaciones potencialmente mortales relacionadas con anomalías cardíacas congénitas, derrames pleurales, infiltrados leucémicos o trombocitopenia), y 10 de los 20 pacientes murieron en el primer mes de vida.
- De los 8 pacientes restantes, ninguno necesitó tratamiento intensivo en el momento del diagnóstico o durante el seguimiento.
- Al cabo de una mediana de seguimiento de 3 años, los 16 pacientes con características mieloproliferativas que no cumplían con los criterios de LMMJ estaban vivos, y ninguno de ellos recibió quimioterapia.
- Variantes del gen CBL. Este gen da origen a CBL, una proteína–ligasa de ubiquitina E3 que participa en la selección de proteínas, en especial de tirosinas–cinasas, para su degradación en los proteasomas. Las variantes del gen CBL se presentan en el 10 % al 15 % de los casos de LMMJ; muchos de estos casos se presentan en niños con variantes germinales de CBL.
Las variantes germinales de CBL producen un trastorno autosómico dominante del desarrollo que a menudo se caracteriza por alteración del crecimiento, retraso del desarrollo, criptorquidia y predisposición a la LMMJ. Algunas personas con variantes germinales de CBL tienen remisión espontánea de la LMMJ pero presentan vasculitis más tarde en su vida, mientras que los pacientes que solo tienen variantes somáticas de CBL necesitan tratamiento. La LMMJ que surge por variantes germinales es indiferenciable (desde el punto de vista clínico) de la LMMJ que surge por variantes somáticas, lo que exige estudios en tejido normal y leucémico. Las variantes de CBL casi siempre son mutuamente excluyentes de las variantes de RAS y PTPN11.
References
- Chan RJ, Cooper T, Kratz CP, et al.: Juvenile myelomonocytic leukemia: a report from the 2nd International JMML Symposium. Leuk Res 33 (3): 355-62, 2009.
- Loh ML: Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol 152 (6): 677-87, 2011.
- Bresolin S, Zecca M, Flotho C, et al.: Gene expression-based classification as an independent predictor of clinical outcome in juvenile myelomonocytic leukemia. J Clin Oncol 28 (11): 1919-27, 2010.
- Olk-Batz C, Poetsch AR, Nöllke P, et al.: Aberrant DNA methylation characterizes juvenile myelomonocytic leukemia with poor outcome. Blood 117 (18): 4871-80, 2011.
- Stiller CA, Chessells JM, Fitchett M: Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer 70 (5): 969-72, 1994.
- Choong K, Freedman MH, Chitayat D, et al.: Juvenile myelomonocytic leukemia and Noonan syndrome. J Pediatr Hematol Oncol 21 (6): 523-7, 1999 Nov-Dec.
- Niemeyer CM, Arico M, Basso G, et al.: Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89 (10): 3534-43, 1997.
- Tartaglia M, Niemeyer CM, Fragale A, et al.: Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34 (2): 148-50, 2003.
- Kratz CP, Niemeyer CM, Castleberry RP, et al.: The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 106 (6): 2183-5, 2005.
- Strullu M, Caye A, Lachenaud J, et al.: Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet 51 (10): 689-97, 2014.
- Loh ML, Sakai DS, Flotho C, et al.: Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 114 (9): 1859-63, 2009.
- Muramatsu H, Makishima H, Jankowska AM, et al.: Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood 115 (10): 1969-75, 2010.
- Niemeyer CM, Kang MW, Shin DH, et al.: Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet 42 (9): 794-800, 2010.
- Pérez B, Mechinaud F, Galambrun C, et al.: Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet 47 (10): 686-91, 2010.
- Hecht A, Meyer JA, Behnert A, et al.: Molecular and phenotypic diversity of CBL-mutated juvenile myelomonocytic leukemia. Haematologica 107 (1): 178-186, 2022.
Características genómicas de la leucemia mielomonocítica juvenil
Características moleculares de la leucemia mielomonocítica juvenil
El panorama genómico de la leucemia mielomonocítica juvenil (LMMJ) se caracteriza por variantes de 1 de los 5 genes de la vía RAS: NF1, NRAS, KRAS, PTPN11 y CBL. En una serie de 118 casos de diagnóstico consecutivo de LMMJ con variantes que activan la vía RAS, PTPN11 fue el gen alterado con mayor frecuencia: representó el 51 % de los casos (19 % en la línea germinal y 32 % somáticos) (consultar la Figura 2). Los pacientes con una variante de NRAS representaron el 19 % de los casos y los pacientes con una variante de KRAS representaron el 15 % de los casos. Las variantes de NF1 representaron el 8 % de los casos y las variantes de CBL representaron el 11 % de los casos. Aunque las variantes de estos 5 genes suelen ser mutuamente excluyentes, el 4 % al 17 % de los casos tienen variantes de 2 de estos genes de la vía RAS, un hallazgo que se relaciona con un pronóstico más precario.
La tasa de variantes de las células leucémicas en la JMML es muy baja, pero se observan variantes adicionales de genes diferentes a los 5 genes de la vía RAS descritos antes. Se observaron alteraciones genómicas secundarias en los genes del complejo represor de la transcripción PRC2 (por ejemplo, ASXL1 estaba alterado en un 7–8 % de los casos). Algunos genes relacionados con neoplasias mieloproliferativas en los adultos también tienen tasas bajas de alteraciones en la JMML (por ejemplo, SETBP1 estaba alterado en un 6–9 % de los casos). También se observaron variantes de JAK3 en un pequeño porcentaje de casos de LMMJ (4–12 %). Los casos con variantes de la línea germinal en PTPN11 y de la línea germinal en CBL exhibieron tasas bajas de variantes adicionales (consultar la Figura 2). La presencia de variantes adicionales a las variantes de la vía RAS, que definen la enfermedad, se relacionan con un pronóstico más precario.
En un informe en el que se describe el panorama genómico de la LMMJ se encontró que 16 de 150 pacientes (11 %) carecían de variantes canónicas de la vía RAS. De esos 16 pacientes, 3 presentaban fusiones en el marco de lectura que afectaban receptores tirosina–cinasas (fusiones génicas DCTN1::ALK, RANBP2::ALK y TBL1XR1::ROS1). Todos estos pacientes presentaban monosomía 7 y tenían 56 meses o más. Un paciente con fusión del gen ALK se trató con crizotinib y quimioterapia convencional, logró una remisión molecular completa, luego se sometió a un trasplante alogénico de médula ósea.
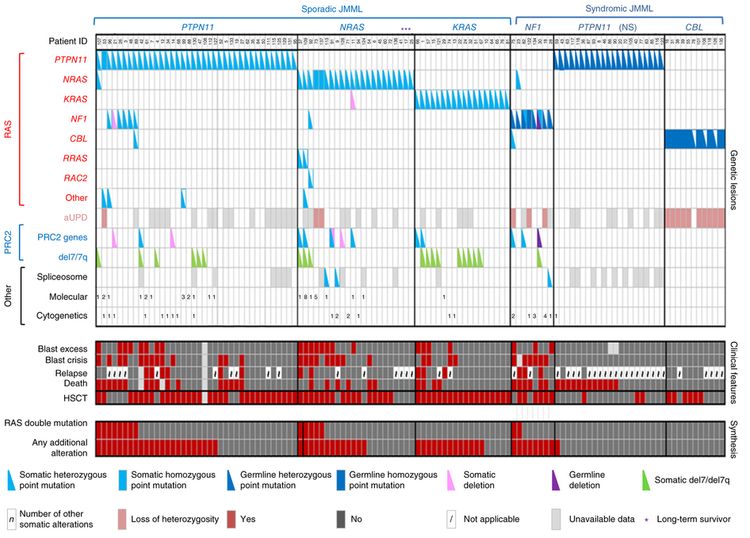 Figura 2. Perfiles de alteraciones en casos individuales de LMMJ. Se muestran las alteraciones de la línea germinal y somáticas con coincidencias recurrentes en la vía RAS y la red PRC2 de 118 pacientes con LMMJ sometidos a pruebas genéticas minuciosas. El exceso de blastocitos se definió como un recuento de blastocitos ≥10 %, pero <20 % de células nucleadas en la médula ósea en el momento del diagnóstico. La crisis blástica se definió como un recuento de blastocitos con ≥20 % de células nucleadas en la médula ósea. NS, síndrome de Noonan. Reproducción autorizada de Macmillan Publishers Ltd: Nature Genetics (Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 [11]: 1334-40, 2015), Derechos de autor (2015). Sporadic JMML: leucemia mielomonocítica juvenil (LMMJ) esporádica; syndromic JMML: LMMJ sindrómica; patient ID: identificación del paciente; other: otro; del: deleción; spliceosome: empalmosoma; cytogenetics: citogenética; blast excess: exceso de blastocitos; blast crisis: crisis blástica; relapse: recaída; death: muerte; HSCT: trasplante de células madre hematopoyéticas (TCMH); RAS double mutation: mutación doble en RAS; any additional alteration: cualquier alteración adicional; somatic heterozygous point mutation: mutación puntual somática heterocigótica; somatic homozygous point mutation: mutación puntual somática homocigótica; germline heterozygous point mutation: mutación puntual en la línea germinal heterocigótica; germline homozygous point mutation: mutación puntual en la línea germinal homocigótica; somatic deletion: deleción somática; germline deletion: deleción de línea germinal; number of other somatic alterations: número de alteraciones somáticas adicionales; loss of heterozygosity: pérdida de heterocigosis; yes: sí; not applicable: no corresponde; unavailable data: no se dispone de datos; long-term survivor: sobreviviente a plazo largo; genetic lessions: lesiones genéticas; clinical features: características clínicas; synthesis: síntesis.
Figura 2. Perfiles de alteraciones en casos individuales de LMMJ. Se muestran las alteraciones de la línea germinal y somáticas con coincidencias recurrentes en la vía RAS y la red PRC2 de 118 pacientes con LMMJ sometidos a pruebas genéticas minuciosas. El exceso de blastocitos se definió como un recuento de blastocitos ≥10 %, pero <20 % de células nucleadas en la médula ósea en el momento del diagnóstico. La crisis blástica se definió como un recuento de blastocitos con ≥20 % de células nucleadas en la médula ósea. NS, síndrome de Noonan. Reproducción autorizada de Macmillan Publishers Ltd: Nature Genetics (Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 [11]: 1334-40, 2015), Derechos de autor (2015). Sporadic JMML: leucemia mielomonocítica juvenil (LMMJ) esporádica; syndromic JMML: LMMJ sindrómica; patient ID: identificación del paciente; other: otro; del: deleción; spliceosome: empalmosoma; cytogenetics: citogenética; blast excess: exceso de blastocitos; blast crisis: crisis blástica; relapse: recaída; death: muerte; HSCT: trasplante de células madre hematopoyéticas (TCMH); RAS double mutation: mutación doble en RAS; any additional alteration: cualquier alteración adicional; somatic heterozygous point mutation: mutación puntual somática heterocigótica; somatic homozygous point mutation: mutación puntual somática homocigótica; germline heterozygous point mutation: mutación puntual en la línea germinal heterocigótica; germline homozygous point mutation: mutación puntual en la línea germinal homocigótica; somatic deletion: deleción somática; germline deletion: deleción de línea germinal; number of other somatic alterations: número de alteraciones somáticas adicionales; loss of heterozygosity: pérdida de heterocigosis; yes: sí; not applicable: no corresponde; unavailable data: no se dispone de datos; long-term survivor: sobreviviente a plazo largo; genetic lessions: lesiones genéticas; clinical features: características clínicas; synthesis: síntesis.
Factores pronósticos genómicos y moleculares
Varios factores genómicos afectan el pronóstico de los pacientes con LMMJ; entre ellos, los siguientes:
- Número de variantes fuera de la vía RAS. Un factor de pronóstico de los niños con LMMJ es el número de variantes diferentes de las variantes de la vía RAS que son definitorias de enfermedad.
- En un estudio se observó que, en el momento del diagnóstico, se encontraron entre 0 a 1 alteraciones somáticas (variante patógena o monosomía 7) en 64 pacientes (65,3 %), mientras que se encontraron 2 o más alteraciones en 34 pacientes (34,7 %). En el análisis multivariante, el número de variantes (2 o más vs. 0 a 1) conservó la significación como factor de pronóstico de supervivencia sin complicaciones (SSC) y supervivencia general (SG) más precarias. Una mayor proporción de pacientes con diagnóstico de 2 o más alteraciones tenían más edad y eran varones; estos pacientes también exhibieron una tasa más alta de monosomía 7 o variantes somáticas de NF1.
- En otro estudio se observó que alrededor del 60 % de los pacientes tenían 1 o más variantes adicionales a la variante de la vía RAS que son definitorias de la enfermedad. Estos pacientes tenían una SG inferior en comparación con los pacientes que no tenían variantes adicionales (tasa de SG a 3 años, 61 vs 85 %, respectivamente).
- En un tercer estudio se observó una tendencia hacia una SG inferior para los pacientes con 2 variantes o más en comparación con los pacientes con 1 o ninguna variante.
- Variantes dobles de la vía RAS. A pesar de que las variantes de los 5 genes canónicos de la vía RAS relacionados con la LMMJ (NF1, NRAS, KRAS, PTPN11, y CBL) son por lo general mutuamente excluyentes, el 4 % al 17 % de los casos presentan variantes de 2 de estos genes de la vía RAS. Este hallazgo se relacionó con un pronóstico más precario.
- En un informe se identificaron 2 variantes de la vía RAS en el 11 % de los pacientes de LMMJ, y estos pacientes tuvieron una tasa de SSC significativamente inferior (14 %) en comparación con la de los pacientes que tenían una sola variante de la vía RAS (62 %). Los pacientes con el síndrome de Noonan se excluyeron del análisis.
- Se informaron resultados similares en un segundo estudio. En este estudio se observó que los pacientes con variantes dobles de la vía RAS (15 de 96 pacientes) tenían tasas de supervivencia más bajas que los pacientes sin variantes adicionales o con variantes adicionales a la variante de la vía RAS.
- Perfil de metilación del DNA.
- En un estudio se aplicó un perfil de metilación del DNA a una cohorte de descubrimiento de 39 pacientes de LMMJ y a una cohorte de validación de 40 pacientes. En ambas cohortes se observaron subgrupos de LMMJ característicos con grados de metilación alto, medio o bajo. Los pacientes con los grados de metilación más bajos tuvieron las tasas de supervivencia más altas, y en la cohorte de metilación baja todos menos 1 de los 15 pacientes presentaron resolución espontánea. El estado de metilación alta se relacionó con tasas más bajas de SSC.
- En otro estudio se aplicó un perfil de metilación del DNA a una cohorte de 106 pacientes de LMMJ. En el estudio se observó un subgrupo de pacientes con un perfil de hipermetilación y un subgrupo de pacientes con un perfil de hipometilación. Los pacientes del grupo de hipermetilación tuvieron una SG significativamente más baja que la de los pacientes del grupo de hipometilación (tasa de SG a 5 años, 46 vs. 73 %, respectivamente). Los pacientes en el grupo de hipermetilación también tuvieron una tasa de supervivencia sin trasplante a 5 años significativamente más precaria que la de los pacientes del grupo de hipometilación (2,2 %, IC 95 %, 0,2–10,1 vs. 41,2 %; IC 95 %, 27,1–54,8 %). El estado de hipermetilación se relacionó con 2 o más variantes, concentraciones más altas de hemoglobina fetal, mayor edad y recuento de plaquetas más bajo en el momento del diagnóstico. Todos los pacientes con síndrome de Noonan estaban en el grupo de hipometilación.
- En un estudio se examinaron 33 pacientes de LMMJ que tenían variantes de CBL. En el estudio se identificaron 31 pacientes con metilación baja y 2 pacientes con metilación intermedia. Los 2 niños con metilación intermedia recayeron después de someterse a TCMH. Debido a que el tratamiento, que incluyó la observación sola, varió entre los 31 pacientes con metilación baja, no se pudo valorar por completo el efecto del perfil de metilación en las decisiones terapéuticas y los desenlaces. Sin embargo, el estado de metilación no fue pronóstico de resolución espontánea.
- Sobreexpresión de LIN28B. La sobreexpresión de LIN28B se presenta en casi la mitad de los niños con LMMJ e identifica un subgrupo de LMMJ distintivo desde el punto de vista biológico. La LIN28B es una proteína de unión al RNA que regula la renovación de células madre.
- La sobreexpresión de LIN28B se correlacionó de forma directa con las concentraciones sanguíneas altas de hemoglobina fetal y la edad (ambos relacionados con un pronóstico más precario), y se correlacionó de forma inversa con monosomía 7 (también relacionada con un pronóstico más precario). Aunque la sobreexpresión de LIN28B permite identificar un subconjunto de pacientes con aumento de riesgo de fracaso del tratamiento, se encontró que no era un factor de pronóstico independiente cuando se consideran otros factores como la edad o la monosomía 7.
- En otro estudio también se observó un subgrupo de pacientes de LMMJ con aumento de la expresión de LIN28B. En el estudio se identificó a LIN28B como el gen cuya expresión se relacionó de manera más contundente con el estado de hipermetilación.
References
- Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 (11): 1334-40, 2015.
- Stieglitz E, Taylor-Weiner AN, Chang TY, et al.: The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47 (11): 1326-33, 2015.
- Murakami N, Okuno Y, Yoshida K, et al.: Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 131 (14): 1576-1586, 2018.
- Sakaguchi H, Okuno Y, Muramatsu H, et al.: Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet 45 (8): 937-41, 2013.
- Stieglitz E, Mazor T, Olshen AB, et al.: Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat Commun 8 (1): 2127, 2017.
- Hecht A, Meyer JA, Behnert A, et al.: Molecular and phenotypic diversity of CBL-mutated juvenile myelomonocytic leukemia. Haematologica 107 (1): 178-186, 2022.
- Helsmoortel HH, Bresolin S, Lammens T, et al.: LIN28B overexpression defines a novel fetal-like subgroup of juvenile myelomonocytic leukemia. Blood 127 (9): 1163-72, 2016.
Factores pronósticos clínicos
En el pasado, más del 90 % de los pacientes con leucemia mielomonocítica juvenil (LMMJ) morían a pesar de la quimioterapia. Sin embargo, con el uso del trasplante de células madre hematopoyéticas, se observan tasas de supervivencia de alrededor del 50 %. Los pacientes siguen tres tipos distintos de evolución clínica:
- Enfermedad rápidamente progresiva y deceso temprano.
- Enfermedad estable transitoria seguida de progresión y muerte.
- Mejoría clínica que dura hasta 9 años antes de la progresión o, de manera infrecuente, supervivencia a largo plazo.
Los factores pronósticos favorables para la supervivencia después de cualquier tratamiento son los siguientes:
- Edad menor de 2 años.
- Recuento de plaquetas superior a 33 × 109/l.
- Concentraciones bajas de hemoglobina fetal ajustadas por edad.
Por el contrario, ser mayor de 2 años y tener concentraciones altas de hemoglobina fetal en sangre en el momento del diagnóstico son factores pronósticos de desenlace precario.
References
- Freedman MH, Estrov Z, Chan HS: Juvenile chronic myelogenous leukemia. Am J Pediatr Hematol Oncol 10 (3): 261-7, 1988 Fall.
- Locatelli F, Nöllke P, Zecca M, et al.: Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood 105 (1): 410-9, 2005.
- Niemeyer CM, Arico M, Basso G, et al.: Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89 (10): 3534-43, 1997.
- Passmore SJ, Chessells JM, Kempski H, et al.: Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia in the UK: a population-based study of incidence and survival. Br J Haematol 121 (5): 758-67, 2003.
Consideraciones especiales para el tratamiento de niños con cáncer
El cáncer en niños y adolescentes es raro, aunque desde 1975 se ha observado un aumento gradual de la incidencia general. Los niños y adolescentes con cáncer se deben derivar a centros médicos que cuenten con equipos multidisciplinarios de especialistas en oncología con experiencia en el tratamiento de los cánceres que se presentan en la niñez y la adolescencia. Este equipo multidisciplinario incorpora la pericia de los siguientes especialistas en pediatría y otros para asegurar que los niños reciban el tratamiento, los cuidados médicos de apoyo y la rehabilitación que les permitan lograr una supervivencia y calidad de vida óptimas:
- Médicos de atención primaria.
- Cirujanos pediatras.
- Patólogos.
- Radioncólogos pediatras.
- Oncólogos y hematólogos pediatras.
- Especialistas en rehabilitación.
- Enfermeros de oncología pediátrica.
- Trabajadores o asistentes sociales.
- Profesionales de la vida infantil.
- Psicólogos.
- Nutricionistas y dietistas.
Para obtener información específica sobre los cuidados médicos de apoyo para niños y adolescentes con cáncer, consultar los resúmenes de Cuidados médicos de apoyo y cuidados paliativos.
La American Academy of Pediatrics estableció pautas para los centros de oncología pediátrica y su función en el tratamiento de los niños y adolescentes con cáncer. En estos centros de oncología pediátrica, se dispone de ensayos clínicos para la mayoría de los tipos de cáncer que se presentan en niños y adolescentes, y se ofrece la oportunidad de participar a la mayoría de los pacientes y familiares. Por lo general, los ensayos clínicos para los niños y adolescentes con cáncer se diseñan a fin de comparar un tratamiento que parece mejor con el tratamiento estándar actual. En otros tipos de ensayos clínicos se prueban tratamientos nuevos cuando no hay un tratamiento estándar para el cáncer que se ha diagnosticado. La mayoría de los avances en la identificación de tratamientos curativos para los cánceres infantiles se lograron mediante ensayos clínicos. Para obtener información sobre los ensayos clínicos en curso, consultar el portal de Internet del NCI.
References
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Wolfson J, Sun CL, Wyatt L, et al.: Adolescents and Young Adults with Acute Lymphoblastic Leukemia and Acute Myeloid Leukemia: Impact of Care at Specialized Cancer Centers on Survival Outcome. Cancer Epidemiol Biomarkers Prev 26 (3): 312-320, 2017.
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed December 15, 2023.
Tratamiento de la leucemia mielomonocítica juvenil
Las opciones de tratamiento de la leucemia mielomonocítica juvenil (LMMJ) son las siguientes:
- Trasplante de células madre hematopoyéticas.
Trasplante de células madre hematopoyéticas
El trasplante de células madre hematopoyéticas (TCMH) ofrece la mejor probabilidad de cura de la LMMJ.
Evidencia (trasplante de células madre hematopoyéticas):
- En un informe reciente del European Working Group on Childhood Myelodysplastic Syndromes llevado a cabo en múltiples centros, se incluyeron 100 receptores de trasplantes tratados con un régimen preparatorio común de busulfano, ciclofosfamida y melfalán, con globulina antitimocítica o sin esta. Los receptores de trasplantes se habían tratado con quimioterapia pretrasplante de diversa intensidad o fármacos diferenciadores, y algunos pacientes se habían sometido a una esplenectomía.
- La tasa de supervivencia sin complicaciones a 5 años fue del 55 % en los niños con LMMJ que recibieron un TCMH de donante emparentado compatible con HLA idéntico y del 49 % en los niños con LMMJ que recibieron TCMH de donantes no emparentados.
- En el análisis multivariante no se observaron efectos en la supervivencia de la quimioterapia similar a la utilizada para la leucemia mieloide aguda (LMA) versus la quimioterapia de dosis bajas o la ausencia de quimioterapia.
- No se observaron efectos en la supervivencia con una esplenectomía anterior al trasplante o diferencias en el tamaño del bazo.
- No se encontraron diferencias en los desenlaces con los donantes emparentados versus los no emparentados.
- Solo la edad mayor de 4 años y el sexo femenino demostraron ser factores pronósticos precarios para el desenlace y para el aumento del riesgo de recaída (riesgo relativo [RR], 2,24 [1,07–4,69]; P = 0,032 para mayor edad; RR, 2,22 [1,09–4,50]; P = 0,028 para las mujeres).
- En un estudio, el trasplante de sangre de cordón umbilical produjo los siguientes resultados:[Nivel de evidencia C2]
- La tasa de supervivencia sin enfermedad a 5 años fue del 44 %.
- Los desenlaces mejoraron en los niños menores de 1,4 años en el momento del diagnóstico, en aquellos con cariotipo sin monosomía 7 y en los que recibieron unidades de sangre de cordón con compatibilidad de HLA 5/6 a 6/6.
- Esto indica que la sangre de cordón umbilical puede proporcionar una fuente adicional de donantes para este grupo de niños.
- En un pequeño número de pacientes también se notificó el uso de regímenes preparatorios de intensidad reducida para disminuir los efectos adversos del trasplante; en general se usa en pacientes que no son aptos para el TCMH mielosupresor.
- El Children's Oncology Group llevó a cabo un ensayo aleatorizado en niños con LMMJ en el que se comparó un régimen de acondicionamiento de intensidad estándar (busulfano, ciclofosfamida y melfalán) con un régimen de intensidad reducida (busulfano y fludarabina).
- Se interrumpió temprano la inscripción en el ensayo cuando en un análisis intermedio se observó una frecuencia más alta de recaída o persistencia de la enfermedad (7 de 9 pacientes) en los niños que recibieron el régimen de intensidad reducida que en aquellos que recibieron el régimen de intensidad estándar (1 de 6 pacientes).
- El Children's Oncology Group llevó a cabo un ensayo aleatorizado en niños con LMMJ en el que se comparó un régimen de acondicionamiento de intensidad estándar (busulfano, ciclofosfamida y melfalán) con un régimen de intensidad reducida (busulfano y fludarabina).
La recidiva de la enfermedad es la causa principal de fracaso del tratamiento en los niños con LMMJ después de un TCMH y se presenta en el 30 % al 40 % de los casos. Si bien la función de las infusiones de linfocitos de donantes es incierta, en los informes se indica que cerca del 50 % de los pacientes con LMMJ en recaída se pueden tratar con éxito con un segundo TCMH.
En un estudio prospectivo, se trató con azacitidina a 4 niños con LMMJ en recaída después de un trasplante de células madre. De los pacientes, 3 respondieron a la azacitidina y pudieron someterse a un segundo trasplante.
La función de la terapia antileucémica convencional en el tratamiento de la LMMJ no está definida. La falta de criterios consensuados de respuesta para la LMMJ complica la determinación de la función de fármacos específicos para el tratamiento de la LMMJ. Algunos fármacos que han demostrado actividad antileucémica contra la LMMJ son el etopósido, la citarabina, las tiopurinas (tioguanina y mercaptopurina), la isotretinoína y los inhibidores de la farnesil–transferasa, pero con ninguno se ha logrado mejorar el desenlace.; [Nivel de evidencia B4]
References
- Smith FO, King R, Nelson G, et al.: Unrelated donor bone marrow transplantation for children with juvenile myelomonocytic leukaemia. Br J Haematol 116 (3): 716-24, 2002.
- Locatelli F, Nöllke P, Zecca M, et al.: Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood 105 (1): 410-9, 2005.
- Yusuf U, Frangoul HA, Gooley TA, et al.: Allogeneic bone marrow transplantation in children with myelodysplastic syndrome or juvenile myelomonocytic leukemia: the Seattle experience. Bone Marrow Transplant 33 (8): 805-14, 2004.
- Baker D, Cole C, Price J, et al.: Allogeneic bone marrow transplantation in juvenile myelomonocytic leukemia without total body irradiation. J Pediatr Hematol Oncol 26 (3): 200-3, 2004.
- Locatelli F, Niemeyer CM: How I treat juvenile myelomonocytic leukemia. Blood 125 (7): 1083-90, 2015.
- Locatelli F, Crotta A, Ruggeri A, et al.: Analysis of risk factors influencing outcomes after cord blood transplantation in children with juvenile myelomonocytic leukemia: a EUROCORD, EBMT, EWOG-MDS, CIBMTR study. Blood 122 (12): 2135-41, 2013.
- Yabe M, Sako M, Yabe H, et al.: A conditioning regimen of busulfan, fludarabine, and melphalan for allogeneic stem cell transplantation in children with juvenile myelomonocytic leukemia. Pediatr Transplant 12 (8): 862-7, 2008.
- Koyama M, Nakano T, Takeshita Y, et al.: Successful treatment of JMML with related bone marrow transplantation after reduced-intensity conditioning. Bone Marrow Transplant 36 (5): 453-4; author reply 454, 2005.
- Dvorak CC, Satwani P, Stieglitz E, et al.: Disease burden and conditioning regimens in ASCT1221, a randomized phase II trial in children with juvenile myelomonocytic leukemia: A Children's Oncology Group study. Pediatr Blood Cancer 65 (7): e27034, 2018.
- Yoshimi A, Bader P, Matthes-Martin S, et al.: Donor leukocyte infusion after hematopoietic stem cell transplantation in patients with juvenile myelomonocytic leukemia. Leukemia 19 (6): 971-7, 2005.
- Yoshimi A, Mohamed M, Bierings M, et al.: Second allogeneic hematopoietic stem cell transplantation (HSCT) results in outcome similar to that of first HSCT for patients with juvenile myelomonocytic leukemia. Leukemia 21 (3): 556-60, 2007.
- Rubio-San-Simón A, van Eijkelenburg NKA, Hoogendijk R, et al.: Azacitidine (Vidaza®) in Pediatric Patients with Relapsed Advanced MDS and JMML: Results of a Phase I/II Study by the ITCC Consortium and the EWOG-MDS Group (Study ITCC-015). Paediatr Drugs 25 (6): 719-728, 2023.
- Bergstraesser E, Hasle H, Rogge T, et al.: Non-hematopoietic stem cell transplantation treatment of juvenile myelomonocytic leukemia: a retrospective analysis and definition of response criteria. Pediatr Blood Cancer 49 (5): 629-33, 2007.
- Castleberry RP, Emanuel PD, Zuckerman KS, et al.: A pilot study of isotretinoin in the treatment of juvenile chronic myelogenous leukemia. N Engl J Med 331 (25): 1680-4, 1994.
- Woods WG, Barnard DR, Alonzo TA, et al.: Prospective study of 90 children requiring treatment for juvenile myelomonocytic leukemia or myelodysplastic syndrome: a report from the Children's Cancer Group. J Clin Oncol 20 (2): 434-40, 2002.
- Loh ML: Childhood myelodysplastic syndrome: focus on the approach to diagnosis and treatment of juvenile myelomonocytic leukemia. Hematology Am Soc Hematol Educ Program 2010: 357-62, 2010.
- Hasle H: Myelodysplastic and myeloproliferative disorders in children. Curr Opin Pediatr 19 (1): 1-8, 2007.
- Stieglitz E, Ward AF, Gerbing RB, et al.: Phase II/III trial of a pre-transplant farnesyl transferase inhibitor in juvenile myelomonocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (4): 629-36, 2015.
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de un ensayo clínico nacional o institucional en curso:
- NCT05849662 (A Phase I/II Study of Trametinib and Azacitidine for Patients With Newly Diagnosed Juvenile Myelomonocytic Leukemia [JMML]): este ensayo clínico evaluará la inocuidad y eficacia de la combinación de trametinib y azacitidina en pacientes con LMMJ.
Actualizaciones más recientes a este resumen (07/05/2024)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Tratamiento de la leucemia mielomonocítica juvenil
Se añadió texto para indicar que, en un estudio prospectivo, se trató con azacitidina a 4 niños con leucemia mielomonocítica juvenil (LMMJ) en recaída después de un trasplante de células madre. De los pacientes, 3 respondieron a la azacitidina y pudieron someterse a un segundo trasplante (se citó a Rubio-San-Simón et al. como referencia 12).
Opciones de tratamiento en evaluación clínica
Se añadió el estudio NCT05849662 como ensayo clínico en curso disponible para pacientes con LMMJ recién diagnosticado.
El Consejo editorial del PDQ sobre el tratamiento pediátrico es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este resumen del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento de la leucemia mielomonocítica juvenil. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El consejo editorial del PDQ sobre el tratamiento pediátrico, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
- Si el artículo se debe analizar en una reunión del consejo.
- Si conviene añadir texto acerca del artículo.
- Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Tratamiento de la leucemia mielomonocítica juvenil son:
- Alan Scott Gamis, MD, MPH (Children's Mercy Hospital)
- Karen J. Marcus, MD, FACR (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Jessica Pollard, MD (Dana-Farber/Boston Children's Cancer and Blood Disorders Center)
- Michael A. Pulsipher, MD (Huntsman Cancer Institute at University of Utah)
- Rachel E. Rau, MD (University of Washington School of Medicine, Seatle Children’s)
- Lewis B. Silverman, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Malcolm A. Smith, MD, PhD (National Cancer Institute)
- Sarah K. Tasian, MD (Children's Hospital of Philadelphia)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El consejo editorial del PDQ sobre el tratamiento pediátrico emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como “En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]”.
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
PDQ® sobre el tratamiento pediátrico. PDQ Tratamiento de la leucemia mielomonocítica juvenil. Bethesda, MD: National Cancer Institute. Actualización:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como “estándar” o “en evaluación clínica”. Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Comuníquese con el Instituto Nacional del Cáncer
Para obtener más información sobre las opciones para comunicarse con el NCI, incluso la dirección de correo electrónico, el número telefónico o el chat, consultar la página del Servicio de Información de Cáncer del Instituto Nacional del Cáncer.