Instituto Nacional del Cáncer
encontrar mi
Fecha de publicación: Nov 8, 2023
Información de expertos y basada en evidencia del síndrome de Birt-Hogg-Dubé, el gen FLCN, la detección, el cuadro clínico inicial, el tratamiento y el pronóstico de este síndrome.
Síndrome de Birt-Hogg-Dubé
Introducción
El síndrome de Birt-Hogg-Dubé (BHD, o enfermedad de Birt-Hogg-Dubé) es un trastorno hamartomatoso de herencia autosómica dominante causado por variantes patogénicas germinales del genFLCN. Birt lo describió por primera vez en 1977, y se caracteriza por hamartomas cutáneos llamados fibrofoliculomas o tricodiscomas. Las características clínicas del síndrome de BHD también abarcan quistes pulmonares, neumotórax espontáneo y tumores renales de varios tipos histológicos. A veces, los pacientes con síndrome de BHD presentan acrocordones, pero no son un hallazgo diagnóstico porque son comunes en la población general.
La gravedad de la enfermedad varía mucho. Las lesiones cutáneas por lo general aparecen durante la 3.ª o 4.ª décadas de vida, y aumentan de tamaño y número con la edad. Los quistes pulmonares suelen ser bilaterales y multifocales. La mayoría de las personas con quistes pulmonares son asintomáticas, pero tienen un riesgo alto de presentar neumotórax espontáneo.
Del 15 % al 30 % de las personas con síndrome de BHD presentarán tumores renales, a menudo bilaterales, multifocales y de crecimiento lento. Los tumores se diagnostican a una mediana de edad de 46 a 50 años. En el BHD, se diagnostica con más frecuencia tumores híbridos oncocíticos (estos tumores tienen características histológicas de oncocitomas y de tipo cromófobo) (50 %), cáncer de células renales de tipo cromófobo (34 %) y oncocitomas (9 %). También se han descrito tumores de células claras y tumores papilares, pero estos representan menos de un 10 % de los tumores renales del síndrome de BHD. Algunas familias presentan tumores renales y neumotórax espontáneo de herencia autosómica dominante sin manifestaciones cutáneas.
Evolución natural
Las características clínicas del síndrome de BHD incluyen fibrofoliculomas o tricodiscomas (tipos de hamartomas cutáneos), quistes pulmonares o antecedentes de neumotórax y tumores renales de varios tipos histológicos. El síndrome de BHD se caracteriza por heterogeneidad fenotípica, y gran variabilidad en la gravedad de la enfermedad entre diferentes miembros de la misma familia y entre diferentes familias. Hasta la fecha, no hay evidencia de aumento del riesgo de cáncer de piel o de transformación maligna de estas lesiones hamartomatosas.
En 2001, en un estudio familiar se observó que los pacientes con diagnóstico clínico de síndrome de BHD eran 7 veces más propensos a presentar tumores renales que los familiares no afectados. Los pacientes con diagnóstico clínico de síndrome de BHD eran también 50 veces más propensos a presentar un neumotórax espontáneo que los familiares sin manifestaciones clínicas. En este estudio se confirmó que los tumores renales y los neumotórax espontáneos son manifestaciones importantes del síndrome de BHD. Si bien los tumores renales relacionados con el síndrome de BHD a veces son de gran malignidad, por lo general son de crecimiento lento. La mayoría de los pacientes que reciben una atención adecuada solo necesitan someterse a una nefrectomía parcial en cada riñón en el transcurso de su vida. Aunque se ha descrito enfermedad metastásica, es rara.
References
- Toro JR, Wei MH, Glenn GM, et al.: BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 45 (6): 321-31, 2008.
- Nickerson ML, Warren MB, Toro JR, et al.: Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell 2 (2): 157-64, 2002.
- Birt AR, Hogg GR, Dubé WJ: Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol 113 (12): 1674-7, 1977.
- Schmidt LS, Linehan WM: Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol 12 (10): 558-69, 2015.
- Boza JC, Trindade EN, Peruzzo J, et al.: Skin manifestations of obesity: a comparative study. J Eur Acad Dermatol Venereol 26 (10): 1220-3, 2012.
- Sanfilippo AM, Barrio V, Kulp-Shorten C, et al.: Common pediatric and adolescent skin conditions. J Pediatr Adolesc Gynecol 16 (5): 269-83, 2003.
- Yosipovitch G, DeVore A, Dawn A: Obesity and the skin: skin physiology and skin manifestations of obesity. J Am Acad Dermatol 56 (6): 901-16; quiz 917-20, 2007.
- Pavlovich CP, Walther MM, Eyler RA, et al.: Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol 26 (12): 1542-52, 2002.
- Benusiglio PR, Giraud S, Deveaux S, et al.: Renal cell tumour characteristics in patients with the Birt-Hogg-Dubé cancer susceptibility syndrome: a retrospective, multicentre study. Orphanet J Rare Dis 9: 163, 2014.
- Shuch B, Vourganti S, Ricketts CJ, et al.: Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 32 (5): 431-7, 2014.
- Graham RB, Nolasco M, Peterlin B, et al.: Nonsense mutations in folliculin presenting as isolated familial spontaneous pneumothorax in adults. Am J Respir Crit Care Med 172 (1): 39-44, 2005.
- Painter JN, Tapanainen H, Somer M, et al.: A 4-bp deletion in the Birt-Hogg-Dubé gene (FLCN) causes dominantly inherited spontaneous pneumothorax. Am J Hum Genet 76 (3): 522-7, 2005.
- Zbar B, Alvord WG, Glenn G, et al.: Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomarkers Prev 11 (4): 393-400, 2002.
- Stamatakis L, Metwalli AR, Middelton LA, et al.: Diagnosis and management of BHD-associated kidney cancer. Fam Cancer 12 (3): 397-402, 2013.
Características genéticas
Gen FLCN
El gen FLCN, es un gen supresor de tumores descubierto recientemente formado por 14 exones que se ubica en el cromosoma 17p11.2. En los pacientes que tienen el síndrome de Birt-Hogg-Dubé (BHD), se han identificado variantes patogénicas del gen FLCN en todos los exones traducidos; y también se han descrito variantes patogénicas intrónicas. El gen FLCN codifica la foliculina (también conocida como proteína FLCN), una fosfoproteína de 64 kDa muy conservada entre especies.
Prevalencia
Se han descrito menos de 1000 familias afectadas por el síndrome de BHD en el mundo. En los estudios se ha notificado estimaciones de prevalencia de entre 1 en 200 000 y 1 en 500 000 para el BHD. En un estudio en el que se analizaron datos de un biobanco poblacional se encontró que es posible que la incidencia de BHD sea de 10 a 100 veces mayor de lo que se pensaba antes. El 89 % de los participantes de este estudio no recibió un diagnóstico de BHD antes de las pruebas genéticas, lo que indica que las manifestaciones clínicas relacionadas con el BHD son a menudo sutiles.
Correlaciones entre genotipo y fenotipo
No se ha establecido una correlación entre variantes específicas del gen FLCN y las manifestaciones renales, pulmonares y cutáneas asociadas con el BHD. Sin embargo, las personas con una deleción en el tramo de policitosina del exón 11 quizás exhiban un riesgo más bajo de presentar cánceres renales relacionados al BHD que las personas con otras variantes de FLCN pero el tamaño de la muestra fue pequeño y esta observación no se replicó en un estudio posterior de la misma institución. A partir de las 3 manifestaciones clínicas principales (fibrofoliculomas o tricodiscomas; quistes pulmonares o neumotórax; y tumores renales), la penetrancia del síndrome de BHD se considera muy elevada. No se ha descrito anticipación en el síndrome de BHD.
References
- Nickerson ML, Warren MB, Toro JR, et al.: Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell 2 (2): 157-64, 2002.
- Schmidt LS, Nickerson ML, Warren MB, et al.: Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet 76 (6): 1023-33, 2005.
- Toro JR, Wei MH, Glenn GM, et al.: BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 45 (6): 321-31, 2008.
- Maffé A, Toschi B, Circo G, et al.: Constitutional FLCN mutations in patients with suspected Birt-Hogg-Dubé syndrome ascertained for non-cutaneous manifestations. Clin Genet 79 (4): 345-54, 2011.
- Kunogi M, Kurihara M, Ikegami TS, et al.: Clinical and genetic spectrum of Birt-Hogg-Dube syndrome patients in whom pneumothorax and/or multiple lung cysts are the presenting feature. J Med Genet 47 (4): 281-7, 2010.
- Rossing M, Albrechtsen A, Skytte AB, et al.: Genetic screening of the FLCN gene identify six novel variants and a Danish founder mutation. J Hum Genet 62 (2): 151-157, 2017.
- Savatt JM, Shimelis H, Moreno-De-Luca A, et al.: Frequency of truncating FLCN variants and Birt-Hogg-Dubé-associated phenotypes in a health care system population. Genet Med 24 (9): 1857-1866, 2022.
- Muller ME, Daccord C, Taffé P, et al.: Prevalence of Birt-Hogg-Dubé Syndrome Determined Through Epidemiological Data on Spontaneous Pneumothorax and Bayes Theorem. Front Med (Lausanne) 8: 631168, 2021.
Aspectos de la biología molecular
La identificación de un segundo golpe o mutación somática en la mayoría de los tumores renales asociados al síndrome de Birt-Hogg-Dubé (BHD) indica con firmeza que el FLCN es un gen supresor de tumores. Se han identificado mutaciones puntuales (variantes) somáticas en el alelo natural de FLCN, además de pérdida de heterocigosidad en el cromosoma 17p, aunque las mutaciones puntuales son el mecanismo más común de inactivación del segundo alelo del gen FLCN.
Todavía no se han aclarado los mecanismos precisos por los que la inactivación del FLCN conduce a la carcinogénesis. Sin embargo, la foliculina, el producto proteínico del gen FLCN, es un componente del sistema sensor de energía celular. La foliculina, en combinación con 1 o 2 proteínas de interacción con foliculina, FNIP1 y FNIP2, interactúa con AMPK. AMPK es un importante nutriente y sensor de energía celular que regula la actividad de mTOR en respuesta a estos estímulos. Además, AMPK fosforila la foliculina y la FNIP1, pero no se entiende bien el significado de esta modificación postraduccional. La interacción con FNIP1 y FNIP2 exige la presencia del dominio C-terminal de FLCN. La mayoría de las variantes de FLCN asociadas con tumores producen una proteína truncada que carece de este dominio C-terminal o desestabilizan la proteína FLCN.
Varios grupos han estudiado los efectos de la pérdida de la foliculina en la actividad de mTOR. Se demostró activación tisular específica de mTORC1 en un modelo de ratón con gen FLCN inactivado específico de riñón. En este modelo, se activaron mTORC1 y mTORC2 en tumores renales que se formaron en ratones con inactivación heterocigota de FLCN y pérdida subsiguiente del alelo natural, lo que indicó que mTOR quizás cumpla una función en el desarrollo de los tumores relacionados con el síndrome de BHD. En un estudio posterior, se indicó que la glucólisis aeróbica aumenta como consecuencia de la inactivación de FLCN. Este cambio glicolítico, aunque moderado, es una consecuencia de la activación constitutiva de AMPK en células con inactivación de FLCN. Se ha demostrado que la activación de AMPK aumenta el factor inducible por hipoxia de tipo 1 (HIF1) que se ha estudiado bien como un activador transcripcional de varios genes necesarios para la glucólisis aeróbica. Se requiere más investigación sobre los mecanismos de la función supresora de tumores del gen FLCN.
References
- Vocke CD, Yang Y, Pavlovich CP, et al.: High frequency of somatic frameshift BHD gene mutations in Birt-Hogg-Dubé-associated renal tumors. J Natl Cancer Inst 97 (12): 931-5, 2005.
- Baba M, Hong SB, Sharma N, et al.: Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A 103 (42): 15552-7, 2006.
- Hasumi H, Baba M, Hong SB, et al.: Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene 415 (1-2): 60-7, 2008.
- Shaw RJ: LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 196 (1): 65-80, 2009.
- Nahorski MS, Reiman A, Lim DH, et al.: Birt Hogg-Dubé syndrome-associated FLCN mutations disrupt protein stability. Hum Mutat 32 (8): 921-9, 2011.
- Baba M, Furihata M, Hong SB, et al.: Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst 100 (2): 140-54, 2008.
- Hasumi Y, Baba M, Ajima R, et al.: Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci U S A 106 (44): 18722-7, 2009.
- Yan M, Gingras MC, Dunlop EA, et al.: The tumor suppressor folliculin regulates AMPK-dependent metabolic transformation. J Clin Invest 124 (6): 2640-50, 2014.
Manifestaciones clínicas
Las 3 características principales del síndrome de Birt-Hogg-Dubé (BHD) son los fibrofoliculomas o tricodiscomas, los quistes pulmonares y el neumotórax espontáneo, y los tumores renales.
Lesiones cutáneas
Las personas con síndrome de BHD suelen presentar múltiples pápulas pequeñas en forma de cúpula, del mismo color de la piel, en la cara, el cuello y la parte superior del tronco. Estas manifestaciones dermatológicas características se llaman fibrofoliculomas o tricodiscomas (hamartoma del folículo piloso). La edad de diagnóstico de las lesiones cutáneas oscila entre 20 y 72 años (mediana de edad, 54 años). Solo una pequeña proporción de portadores de variantes patogénicas de FLCN carecen de manifestaciones cutáneas, lo que indica que este fenotipo sindrómico es muy penetrante en las personas afectadas. En 2 estudios familiares grandes sobre el síndrome de BHD, se encontró que el 73 % y el 84 % de los pacientes afectados en quienes que se obtuvieron biopsias de lesiones cutáneas tenían fibrofoliculomas o tricodiscomas.
Desde el punto de vista histológico, los fibrofoliculomas o tricodiscomas se caracterizan por múltiples hebras epiteliales anastomosantes que emanan de un folículo central. A veces el estroma está formado por tejido conjuntivo denso o rico en mucina y encapsula el componente epitelial. Algunos las describen como lesiones que emanan de la capa sebácea del folículo piloso. No está claro el mecanismo molecular subyacente, que se deriva de la pérdida de FLCN y que impulsa la formación de los fibrofoliculomas o tricodiscomas, pero en un informe se indica que quizás se debe al aumento de la señalización de WNT. Los fibrofoliculomas y los tricodiscomas son estadios diferentes de un proceso patológico único.
Quistes pulmonares y neumotórax espontáneo
En tomografía computarizada (TC) se observa que hay quistes pulmonares en el 85 % al 87 % de los pacientes con síndrome de BHD. Estos quistes a menudo son bilaterales, multifocales y se ubican de manera predominante en los lóbulos inferiores del pulmón. La mayoría de los quistes de pulmón relacionados con el síndrome de BHD son asintomáticos; sin embargo, este síndrome acarrea aumento del riesgo de neumotórax espontáneo en las personas afectadas. Los pacientes con una variante patogénica de FLCN y una historia familiar de neumotórax espontáneo presentaron un aumento estadísticamente significativo en el riesgo de neumotórax espontáneo en comparación con los pacientes afectados por el síndrome de BHD sin historia familiar de neumotórax espontáneo (P = 0,011).
En un estudio de 198 pacientes con BHD, los casos de neumotórax espontáneo fueron comparables entre hombres (20 %) y mujeres (29 %). Los primero neumotórax se presentaron entre los 22 y los 75 años. Sin embargo, la mediana de edad para el primer caso de neumotórax fue de 38 años y por lo general ocurrió antes de la 5.° década de la vida. Los pacientes tuvieron una probabilidad del 6 % de presentar el primer neumotórax espontáneo a los 30 años (intervalo de confianza [IC] de 95 %, 3–10 %). Esta probabilidad aumentó hasta el 75 % a los 50 años (IC 95 %, 19–32 %). En una revisión de publicaciones de PubMed en inglés se analizaron datos de 1038 pacientes con BHD y se encontró que el 50,9 % de ellos presentó neumotórax. La incidencia variaba según la ascendencia de la persona.
El cuadro clínico del neumotórax espontáneo varía desde ausencia de síntomas hasta disnea y dolor torácico. Los hallazgos clínicos incluyen taquipnea o murmullo respiratorio disminuido o ausente. Es posible que la investigación radiográfica exija el uso de una TC torácica de alta resolución para confirmar el diagnóstico porque la radiografía del tórax quizás no sea lo suficientemente sensible como para detectar un neumotórax loculado. Hasta el 75 % de los pacientes con antecedentes de neumotórax espontáneo presentarán un segundo caso de neumotórax espontáneo. Las diferencias en la notificación de la recidiva del neumotórax espontáneo quizás sean un reflejo de la eficacia de las diferentes modalidades de tratamiento.
Los hallazgos histológicos de las lesiones pleuropulmonares relacionadas con el síndrome de BHD incluyen bullas y quistes pleurales y subpleurales de pared delgada, quistes de aire intraparenquimatosos, vesículas pleurales y cambios compatibles con neumotórax espontáneo, además de cambios enfisematosos subyacentes en el parénquima pulmonar adyacente a las bullas.
Tumores renales
Del 25 % al 35 % de las personas afectadas por el síndrome de BHD presentarán tumores renales, que son multifocales en el 65 % de los casos y, a menudo, bilaterales. La frecuencia de tumores renales en los pacientes con síndrome de BHD fue del 20 % según la revisión de historias clínicas y del 29 % según la evaluación mediante TC. La mayoría de los tumores renales relacionados con el síndrome de BHD son de crecimiento lento. La mediana de edad en el momento del diagnóstico oscila entre 48 y 50 años (intervalo, 31–71 años). Los tumores renales fueron más frecuentes en los hombres que en las mujeres (27 hombres; 11 mujeres). Los tumores renales relacionados con el síndrome de BHD surgen a una edad más temprana que las formas esporádicas del cáncer de células renales (RCC), cuya mediana de edad es de 64 años en el momento del diagnóstico. En la Figura 1 se observan tumores renales bilaterales en un paciente con síndrome de BHD.
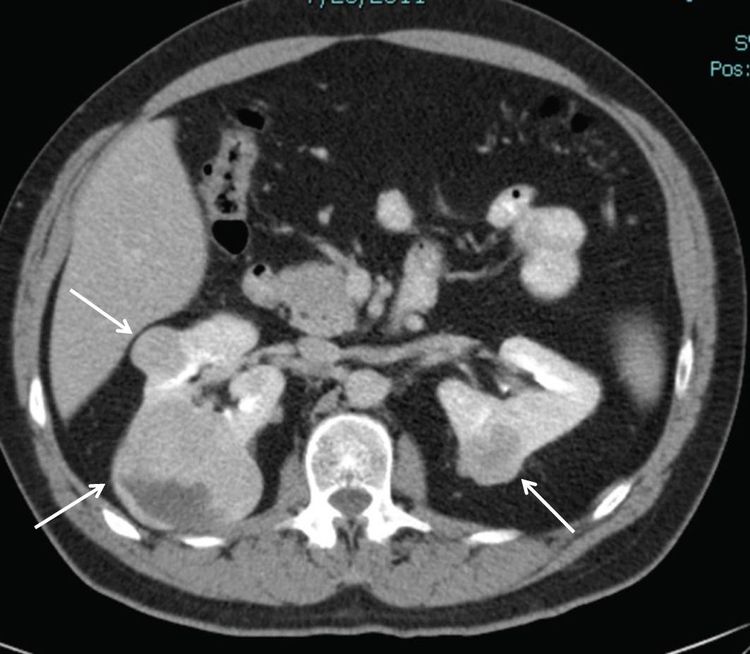 Figura 1. Los tumores renales asociados con el síndrome de Birt-Hogg-Dubé suelen ser multifocales y bilaterales. Las flechas señalan la ubicación de los tumores.
Figura 1. Los tumores renales asociados con el síndrome de Birt-Hogg-Dubé suelen ser multifocales y bilaterales. Las flechas señalan la ubicación de los tumores.
Los tumores más comunes son un híbrido de los tipos histológicos celulares oncocitoma y cromófobo, los llamados tumores híbridos oncocíticos, el carcinoma de células renales cromófobo y el oncocitoma renal. Solo el oncocitoma renal se considera un tumor benigno. Otros subtipos histológicos de tumor renal, como el cáncer renal de células claras (ccRCC) y el carcinoma papilar renal, son poco frecuentes en los pacientes afectados por el síndrome de BHD.
Entre 70 pacientes con síndrome de BHD, tumores renales y una variante patogénica de FLCN analizados por los Institutos Nacionales de la Salud e identificados mediante revisión de la literatura, se notificó que 5 (7 %) fallecieron debido a RCC metastásico. El tipo histológico tumoral en estos 5 pacientes abarcó células claras, y características tubulopapilares o papilares, que se sabe exhiben una evolución natural biológica más maligna. La muerte relacionada con el oncocitoma y las neoplasias cromófobas propias del síndrome de BHD es muy infrecuente. De manera similar a la enfermedad de Von Hippel-Lindau y al carcinoma renal papilar hereditario, el parénquima renal de los pacientes con síndrome de BHD suele mostrar tumores renales microscópicos adyacentes a los carcinomas de células renales. La presencia de oncocitosis microscópica proporciona indicios histológicos de que el síndrome de BHD acarrea riesgo de tumores renales durante toda la vida. La frecuencia alta de «segundos golpes» somáticos en FLCN (70 %) en los tumores renales asociados con el síndrome de BHD respalda la hipótesis de que el FLCN actúa como un gen supresor de tumores. Solo en algunos pocos casos de ccRCC esporádico se han detectado mutaciones somáticas, es decir adquiridas, en FLCN.
Otras manifestaciones
Se notificaron oncocitomas de parótida multifocales bilaterales en 8 pacientes con síndrome de BHD. La presentación multifocal bilateral de estos tumores raros, en combinación con investigaciones moleculares recientes, ha llevado a especular que los oncocitomas de parótida podrían formar parte de la variedad fenotípica del síndrome de BHD.
Cabe señalar que también se encontraron variantesgerminales de FLCN en pacientes sin hallazgos cutáneos, pero con sospecha de síndrome de BHD debido a manifestaciones renales y pulmonares específicas.
Además, se han notificado lipomas, angiolipomas, colagenomas, neurotequeomas cutáneos, meningiomas, bocios multinodulares de tiroides, quistes de ovario, adenomas de paratiroides, histiocitomas pulmonares y lesiones coriorretinianas en pacientes con síndrome de BHD. Queda por determinar si estas manifestaciones están verdaderamente relacionadas con el síndrome de BHD.
Aunque las observaciones epidemiológicas iniciales permitieron establecer un vínculo entre el síndrome de BHD y un aumento del riesgo de pólipos en el colon, en estudios epidemiológicos posteriores no se confirmó esta asociación. En un estudio que se realizó en los Países Bajos, las personas con BHD no presentaron una mayor prevalencia de cáncer colorrectal (CCR) en comparación con sus familiares sin esta afección, a pesar de que para las personas con BHD se usaron pruebas de detección del CCR ampliadas.
References
- Toro JR, Wei MH, Glenn GM, et al.: BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 45 (6): 321-31, 2008.
- Schmidt LS, Nickerson ML, Warren MB, et al.: Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet 76 (6): 1023-33, 2005.
- Schmidt LS, Linehan WM: Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol 12 (10): 558-69, 2015.
- Toro JR, Glenn G, Duray P, et al.: Birt-Hogg-Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol 135 (10): 1195-202, 1999.
- Graham RB, Nolasco M, Peterlin B, et al.: Nonsense mutations in folliculin presenting as isolated familial spontaneous pneumothorax in adults. Am J Respir Crit Care Med 172 (1): 39-44, 2005.
- Painter JN, Tapanainen H, Somer M, et al.: A 4-bp deletion in the Birt-Hogg-Dubé gene (FLCN) causes dominantly inherited spontaneous pneumothorax. Am J Hum Genet 76 (3): 522-7, 2005.
- Vernooij M, Claessens T, Luijten M, et al.: Birt-Hogg-Dubé syndrome and the skin. Fam Cancer 12 (3): 381-5, 2013.
- Toro JR, Pautler SE, Stewart L, et al.: Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dubé syndrome. Am J Respir Crit Care Med 175 (10): 1044-53, 2007.
- Matsumoto K, Lim D, Pharoah PD, et al.: A systematic review assessing the existence of pneumothorax-only variants of FLCN. Implications for lifelong surveillance of renal tumours. Eur J Hum Genet 29 (11): 1595-1600, 2021.
- Zbar B, Alvord WG, Glenn G, et al.: Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomarkers Prev 11 (4): 393-400, 2002.
- Pavlovich CP, Walther MM, Eyler RA, et al.: Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol 26 (12): 1542-52, 2002.
- Pavlovich CP, Grubb RL, Hurley K, et al.: Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J Urol 173 (5): 1482-6, 2005.
- Shuch B, Vourganti S, Ricketts CJ, et al.: Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 32 (5): 431-7, 2014.
- Sattler EC, Reithmair M, Steinlein OK: Kidney cancer characteristics and genotype-phenotype-correlations in Birt-Hogg-Dubé syndrome. PLoS One 13 (12): e0209504, 2018.
- Howlader N, Noone AM, Krapcho M, et al.: SEER Cancer Statistics Review (CSR) 1975-2017. Bethesda, Md: National Cancer Institute, 2020. Available online. Last accessed January 26, 2024.
- Vocke CD, Yang Y, Pavlovich CP, et al.: High frequency of somatic frameshift BHD gene mutations in Birt-Hogg-Dubé-associated renal tumors. J Natl Cancer Inst 97 (12): 931-5, 2005.
- da Silva NF, Gentle D, Hesson LB, et al.: Analysis of the Birt-Hogg-Dubé (BHD) tumour suppressor gene in sporadic renal cell carcinoma and colorectal cancer. J Med Genet 40 (11): 820-4, 2003.
- Khoo SK, Kahnoski K, Sugimura J, et al.: Inactivation of BHD in sporadic renal tumors. Cancer Res 63 (15): 4583-7, 2003.
- Liu V, Kwan T, Page EH: Parotid oncocytoma in the Birt-Hogg-Dubé syndrome. J Am Acad Dermatol 43 (6): 1120-2, 2000.
- Vinit J, Friedel J, Bielefeld P, et al.: [Birt-Hogg-Dubé syndrome and multiple recurrent tumors]. Rev Med Interne 32 (3): e40-2, 2011.
- Maffé A, Toschi B, Circo G, et al.: Constitutional FLCN mutations in patients with suspected Birt-Hogg-Dubé syndrome ascertained for non-cutaneous manifestations. Clin Genet 79 (4): 345-54, 2011.
- Chung JY, Ramos-Caro FA, Beers B, et al.: Multiple lipomas, angiolipomas, and parathyroid adenomas in a patient with Birt-Hogg-Dube syndrome. Int J Dermatol 35 (5): 365-7, 1996.
- Vincent A, Farley M, Chan E, et al.: Birt-Hogg-Dubé syndrome: two patients with neural tissue tumors. J Am Acad Dermatol 49 (4): 717-9, 2003.
- Drummond C, Grigoris I, Dutta B: Birt-Hogg-Dubé syndrome and multinodular goitre. Australas J Dermatol 43 (4): 301-4, 2002.
- Welsch MJ, Krunic A, Medenica MM: Birt-Hogg-Dubé Syndrome. Int J Dermatol 44 (8): 668-73, 2005.
- Tomassetti S, Carloni A, Chilosi M, et al.: Pulmonary features of Birt-Hogg-Dubé syndrome: cystic lesions and pulmonary histiocytoma. Respir Med 105 (5): 768-74, 2011.
- Walter P, Kirchhof B, Korge B, et al.: Flecked chorioretinopathy associated with Birt-Hogg-Dubé syndrome. Graefes Arch Clin Exp Ophthalmol 235 (6): 359-61, 1997.
- Nahorski MS, Lim DH, Martin L, et al.: Investigation of the Birt-Hogg-Dube tumour suppressor gene (FLCN) in familial and sporadic colorectal cancer. J Med Genet 47 (6): 385-90, 2010.
- van de Beek I, Glykofridis IE, Wolthuis RMF, et al.: No evidence for increased prevalence of colorectal carcinoma in 399 Dutch patients with Birt-Hogg-Dubé syndrome. Br J Cancer 122 (4): 590-594, 2020.
Atención médica
Evaluación del riesgo del síndrome de Birt-Hogg-Dubé
Pruebas genéticas
El FLCN es el único gen del que se conoce su asociación con el síndrome de Birt-Hogg-Dubé (BHD). Está ubicado en el cromosoma 17p11.2. Hay pruebas moleculares de aplicación clínica como las pruebas diagnósticas y de diagnóstico prenatal. De las familias con síndrome de BHD, el 53 % (27 de 51) tenían una inserción o deleción en el tramo de policitosina en el exón 11 (punto caliente de variantes). La secuenciación bidireccional del DNA de todos los exones codificadores de FLCN (exón 4–14) derivaron en una tasa de detección de variantes patogénicas del 84 %. Esta tasa ha mejorado aún más con el desarrollo de las pruebas de reacción en cadena de la polimerasa cuantitativas ultrarrápidas y amplificación múltiple de sondas dependiente de ligamiento para detectar duplicaciones y deleciones intragénicas; estas pruebas están disponibles en el entorno clínico.
Se indica el uso de pruebas genéticas a cargo de laboratorios con certificación según las Enmiendas para el Mejoramiento de Laboratorios Clínicos (CLIA) para todas las personas con un presunto síndrome de BHD o en quienes ya se confirmó la afectación, entre ellos quienes presenten las siguientes características:
- 5 o más pápulas en la cara o el tronco, y por lo menos 1 fibrofoliculoma confirmado mediante análisis histológico, con historia familiar compatible con síndrome de BHD o sin estos antecedentes familiares.
- Historia familiar compatible con el síndrome de BHD y un fibrofoliculoma único, un tumor renal único o antecedentes personales de neumotórax espontáneo.
- Tumores cromófobos múltiples y bilaterales, o tumores renales híbridos oncocíticos.
- 1 solo tumor cromófobo o 1 tumor híbrido oncocítico acompañado de una historia familiar compatible con cáncer renal de cualquiera de los tipos de células renales mencionados antes.
- Una historia familiar de neumotórax espontáneo primario de herencia autosómica dominante sin antecedentes de quistes pulmonares.
Asesoramiento genético
El síndrome de BHD se hereda de manera autosómica dominante. Si un progenitor de un probando está afectado clínicamente o tiene una variante patogénica de FLCN , los hermanos o hermanas del probando tienen una probabilidad del 50 % de heredar la variante. La gravedad clínica no es predecible. El diagnóstico prenatal de BHD es posible durante el embarazo con un 50 % de probabilidad de heredar una variante patogénica de FLCN si la variante que causa la enfermedad se ha identificado en un miembro de la familia afectado.
En el resumen Evaluación del riesgo de cáncer y asesoramiento genético se proporciona más información acerca de estos temas:
- Para obtener más información sobre el diagnóstico prenatal de los síndromes de cáncer hereditario, consultar la sección Pruebas genéticas y técnicas de reproducción asistida.
- Para obtener más información sobre la herencia autosómica dominante, consultar la sección Análisis de la historia familiar.
- Para obtener más información sobre las pruebas genéticas para detectar variantes patogénicas en miembros de una familia, consultar la sección Pruebas genéticas en cascada en los miembros de una familia.
Diagnóstico clínico
Las 3 características principales del síndrome de BHD son las lesiones cutáneas, las manifestaciones pulmonares (quistes en el pulmón y neumotórax espontáneo) y los tumores renales. Para obtener más información sobre estas manifestaciones, consultar la sección Manifestaciones clínicas en este resumen.
El diagnóstico dermatológico del síndrome de BHD se establece en personas que tienen 5 o más pápulas en la cara o el tronco, y por lo menos 1 fibrofoliculoma confirmado por análisis histológico. Se requiere una biopsia adecuada (por lo general, una biopsia con sacabocados) para determinar el diagnóstico de fibrofoliculoma. Un grupo de expertos planteó los siguientes criterios diagnósticos para el síndrome de BHD (los pacientes deben cumplir con 1 criterio principal o 2 criterios secundarios para determinar el diagnóstico):
- Criterios principales
- Por lo menos 5 fibrofoliculomas o tricodiscomas (al menos 1 confirmado por análisis histológico), y de inicio en la edad adulta.
- Variante patogénica germinal de FLCN.
- Criterios secundarios
- Quistes pulmonares múltiples: quistes pulmonares bilaterales y basales sin otra causa aparente, con neumotórax primario espontáneo o sin este.
- Cáncer renal: cáncer renal multifocal, bilateral o de inicio temprano (edad <50 años), o cáncer renal de tipo histológico mixto cromófobo y oncocítico.
- Un familiar de primer grado con síndrome de BHD.
Diagnóstico diferencial
Es importante distinguir entre el carcinoma de células renales propio del síndrome de BHD y el carcinoma de células renales esporádico porque esto quizás repercuta en el tratamiento. El uso de pruebas genéticas para detectar una variante patogénica en FLCN, una historia familiar de BHD, o la presencia de manifestaciones extrarrenales del BHD son útiles para establecer el diagnóstico de esta afección. Debido a que se pueden observar variantes histológicas del cáncer de riñón asociadas al síndrome de BHD, a menudo es necesario determinar el diagnóstico histológico para ayudar a diferenciar entre tumores benignos (oncocitomas) y los que tienen potencial maligno (carcinoma de células renales cromófobo, de células claras y papilar).
El diagnóstico diferencial de los quistes pulmonares incluye la linfangioleiomiomatosis (LAM); distinguir la LAM del síndrome de BHD es un reto clínico. En un estudio se propuso un conjunto de hallazgos que permiten diferenciar el síndrome de BHD y la LAM. Estos abarcan la distribución bibasal, periférica y subpleural de las lesiones del síndrome de BHD versus la distribución difusa de las lesiones de la LAM; la forma elíptica o lenticular de los quistes relacionados con el síndrome de BHD versus la forma redondeada de los quistes de la LAM; y la negatividad para HMB-45 en la tinción inmunohistoquímica del síndrome de BHD versus la positividad para HMB-45 en casos de LAM. Este abordaje no se ha validado; se justifica realizar más estudios.
Vigilancia
Los pacientes con síndrome de BHD exhiben 2 cuadros clínicos iniciales principales. Con mayor frecuencia, las personas presentan una historia familiar documentada de síndrome de BHD. Otro cuadro clínico inicial ocurre en personas sin una historia familiar compatible con el síndrome de BHD o que desconocen su historia familiar. En el primer escenario clínico, si el paciente tiene un familiar biológico en el que se estableció un diagnóstico genético y en quien se identificó una variante patogénica de FLCN, es posible que el paciente decida comenzar la evaluación con el asesoramiento genético y las pruebas de la variante patogénica.
La vigilancia clínica para las personas en riesgo de BHD incluye exámenes dermatológicos, radiológicos e histológicos para identificar lesiones cutáneas características, tumores renales y quistes pulmonares, con o sin antecedentes de neumotórax espontáneo. No todas las personas en riesgo exhiben todas las características, y es posible que algunos familiares no presenten hallazgos fenotípicos discernibles (es decir, son portadores de variantes de FLCNdeletéreasno afectados desde el punto de vista clínico). Este escenario clínico es cada vez más frecuente a medida que aumenta el número de genes relacionados con el síndrome para los que se pueden ofrecer pruebas de variantes patogénicas en el ámbito clínico. En la mayoría de los trastornos, todavía no se ha caracterizado bien la evolución natural en las personas con anomalías genéticas que son normales desde el punto de vista clínico. Estas características principales del BHD se describen en la sección Diagnóstico clínico.
Las decisiones sobre la vigilancia de por vida para los síndromes hereditarios de carcinoma de células renales deben tener en cuenta tanto los riesgos como los beneficios. Del 15 % al 29 % de las personas afectadas por el síndrome de BHD presentarán tumores renales, que a menudo son bilaterales y multifocales, y abarcan un número específico de tipos histológicos en la misma persona o en la misma familia. Se indica el uso de un programa de vigilancia que reduzca al mínimo la dosis de radiación para las personas en riesgo que se someten a pruebas con imágenes durante muchos años, incluso cuando no hay tumor.
La tomografía computarizada (TC) contrastada o las imágenes por resonancia magnética (IRM) son modalidades útiles para la detección de los tumores renales asociados con el síndrome de BHD. Es posible que una ecografía sola no sea suficiente para la detección de los tumores renales porque algunos tumores son isoecoicos con el parénquima renal, pero es útil para identificar quistes renales. En una serie de los Países Bajos, con la ecografía sola no se lograron detectar 9 de 18 tumores renales. Por lo tanto, aunque la ecografía quizás permita la detección confiable de lesiones más grandes, no es confiable para detectar lesiones más pequeñas y por lo tanto no se debe usar de rutina como el único instrumento de detección. Si se detecta un tumor renal, el paciente se remite para que lo atienda un cirujano especialista en urología oncológica, lo que a veces abarca continuación de la vigilancia o cirugía, según el tamaño del tumor. Si no se detectan tumores renales en las imágenes iniciales, los expertos recomiendan la vigilancia de por vida con citas al menos cada 36 meses debido al riesgo de carcinoma de células renales. Debido a que la IRM evita la exposición del paciente a radiación, es razonable asumir que es el método preferible para la vigilancia de por vida en comparación con la TC.
Nivel de evidencia: 5
Tratamiento
Cutáneo
Se ha usado crioterapia, electrodesecación, cirugía y terapia láser con buenos resultados cosméticos, pero a menudo hay recaída porque las lesiones son una manifestación de una afección cutánea hereditaria. Por lo tanto, es posible que los pacientes requieran atención cosmética continua. Algunos pacientes con BHD se ven afectados emocionalmente por su afección dermatológica, independientemente del número o la extensión de las lesiones cutáneas. Por lo tanto, el estado psicológico de los pacientes con BHD merece consideración, con recomendaciones de cuidado de la piel adaptadas a las necesidades individuales.
Nivel de evidencia: 5
Renal
La nefrectomía parcial es el tratamiento de elección para el abordaje de las neoplasias de riñón relacionadas con el síndrome de BHD, con el fin de conservar el funcionamiento renal óptimo a largo plazo en pacientes con riesgo de múltiples tumores renales primarios. Sin embargo, la cirugía con conservación renal depende del tamaño y la ubicación de los tumores que se encuentran durante la cirugía. Cuando se planifica el tratamiento quirúrgico, es importante incorporar el conocimiento del riesgo acumulado alto de tumores renales multifocales y bilaterales en este síndrome. En general, los tumores renales de menos de 3 cm de diámetro los puede vigilar de cerca el cirujano especialista en urología oncológica y es posible que no se requiera una cirugía inmediata. Estas son recomendaciones generales, pero cada caso se debe evaluar cuidadosamente y el tratamiento debe ser personalizado. En algunos casos, es posible que se necesite una nefrectomía total.
La vigilancia de personas y familiares en riesgo incluye IRM o TC abdominales o pélvicas, además de la evaluación de tumores renales a cargo de urólogos cirujanos y radiólogos con experiencia en el tratamiento de estos pacientes complicados. El uso de una prueba genética para la identificación temprana de los familiares en riesgo mejora la certeza diagnóstica y elimina los costosos y estresantes procedimientos de detección en los familiares en riesgo que no heredaron la variante causal de enfermedad presente en la familia.
Nivel de evidencia: 4
Neumotórax espontáneo
El tratamiento del neumotórax espontáneo en pacientes con el síndrome de BHD es similar al de la población general.
El cuadro clínico inicial del neumotórax espontáneo en pacientes con síndrome de BHD es variable. El tratamiento se determina a partir de la afección pulmonar subyacente y el estado general de salud del paciente. En un estudio se notificó que de 101 pacientes con neumotórax espontáneo, 78 necesitaron una intervención médica, y 23 se abordaron solo con observación. El 35 % de los pacientes con neumotórax se trataron solo con toracostomía cerrada (tubo de drenaje torácico); el 14 % se trató con toracotomía abierta y un segundo tratamiento, como pleurodesis mecánica y química o resección pulmonar. Casi un 13 % se trató con toracostomía cerrada, toracotomía y un tercer tratamiento, como pleurodesis mecánica o química y resección pulmonar. Se debe recomendar a los pacientes con síndrome de BHD, en especial a los que tienen varios quistes pulmonares, que eviten o sean precavidos con el buceo, el transporte aéreo y la ventilación mecánica porque cada exposición aumenta el riesgo de neumotórax espontáneo.
Nivel de evidencia: 4
References
- Schmidt LS, Warren MB, Nickerson ML, et al.: Birt-Hogg-Dubé syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet 69 (4): 876-82, 2001.
- Schmidt LS, Nickerson ML, Warren MB, et al.: Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet 76 (6): 1023-33, 2005.
- Toro JR, Wei MH, Glenn GM, et al.: BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 45 (6): 321-31, 2008.
- Benhammou JN, Vocke CD, Santani A, et al.: Identification of intragenic deletions and duplication in the FLCN gene in Birt-Hogg-Dubé syndrome. Genes Chromosomes Cancer 50 (6): 466-77, 2011.
- Toro JR, Glenn G, Duray P, et al.: Birt-Hogg-Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol 135 (10): 1195-202, 1999.
- Menko FH, van Steensel MA, Giraud S, et al.: Birt-Hogg-Dubé syndrome: diagnosis and management. Lancet Oncol 10 (12): 1199-206, 2009.
- Pavlovich CP, Grubb RL, Hurley K, et al.: Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J Urol 173 (5): 1482-6, 2005.
- Gupta N, Seyama K, McCormack FX: Pulmonary manifestations of Birt-Hogg-Dubé syndrome. Fam Cancer 12 (3): 387-96, 2013.
- Zbar B, Alvord WG, Glenn G, et al.: Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomarkers Prev 11 (4): 393-400, 2002.
- Stamatakis L, Metwalli AR, Middelton LA, et al.: Diagnosis and management of BHD-associated kidney cancer. Fam Cancer 12 (3): 397-402, 2013.
- Johannesma PC, van de Beek I, van der Wel TJWT, et al.: Renal imaging in 199 Dutch patients with Birt-Hogg-Dubé syndrome: Screening compliance and outcome. PLoS One 14 (3): e0212952, 2019.
- Jacob CI, Dover JS: Birt-Hogg-Dube syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol 137 (1): 98-9, 2001.
- Gambichler T, Wolter M, Altmeyer P, et al.: Treatment of Birt-Hogg-Dubé syndrome with erbium:YAG laser. J Am Acad Dermatol 43 (5 Pt 1): 856-8, 2000.
- Kahle B, Hellwig S, Schulz T: [Multiple mantleomas in Birt-Hogg-Dubé syndrome: successful therapy with CO2 laser] Hautarzt 52 (1): 43-6, 2001.
- Toro JR, Pautler SE, Stewart L, et al.: Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dubé syndrome. Am J Respir Crit Care Med 175 (10): 1044-53, 2007.
Pronóstico
La causa principal de morbilidad y mortalidad en el síndrome de Birt-Hogg-Dubé (BHD) está relacionada con las lesiones renales. Debido a la rareza del síndrome de BHD, es difícil generar datos sólidos de supervivencia general en poblaciones de pacientes con este síndrome; sin embargo, cuando los pacientes se tratan con una estrategia apropiada de vigilancia e intervención, su esperanza de vida no debe ser significativamente diferente a la de las personas de la población general emparejadas.
Si bien la mayoría de los pacientes obtienen excelentes resultados cuando los tumores se detectan temprano y se extirpan mediante cirugía, hay riesgo de metástasis con tumores más grandes y el tratamiento óptimo de la enfermedad metastásica no está claro.
References
- Johannesma PC, van de Beek I, van der Wel TJWT, et al.: Renal imaging in 199 Dutch patients with Birt-Hogg-Dubé syndrome: Screening compliance and outcome. PLoS One 14 (3): e0212952, 2019.
Indicaciones futuras
Desde que en 2001 se identificó el genFLCN, responsable del síndrome de Birt-Hogg-Dubé, en varios estudios se ha aclarado su función y las posibles correlaciones entre genotipo y fenotipo. Aunque la vigilancia seguida de resección quirúrgica sigue siendo el pilar del abordaje de la enfermedad, las mejoras en la detección temprana y en las intervenciones moleculares tempranas quizás modifiquen la evolución de esta enfermedad en el riñón al disminuir la incidencia de las manifestaciones renales evidentes o letales. Una mejor comprensión del funcionamiento bioquímico de la proteína FLCN debe proporcionar información sobre la identificación y validación del tratamiento médico para la enfermedad localizada, localmente avanzada y metastásica.
Actualizaciones más recientes a este resumen (11/09/2023)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Características genéticas
Se revisó el texto para indicar que menos de 1000 familias afectadas por el síndrome de Birt-Hogg-Dubé (BHD) en el mundo (se citó a Savatt et al. como referencia 7 y Muller et al. como referencia 8). En los estudios se ha notificado estimaciones de prevalencia de entre 1 en 200 000 y 1 en 500 000 para el BHD. En un estudio en el que se analizaron datos de un biobanco poblacional se encontró que es posible que la incidencia de BHD sea de 10 a 100 veces mayor de lo que se pensaba antes. El 89 % de los participantes de este estudio no recibió un diagnóstico de BHD antes de las pruebas genéticas, lo que indica que las manifestaciones clínicas relacionadas con el BHD son a menudo sutiles.
Manifestaciones clínicas
Se añadió texto para indicar que en una revisión de publicaciones de la de PubMed en inglés se analizaron datos de 1038 pacientes con BHD y se encontró que el 50,9 % de ellos presentó neumotórax. La incidencia variaba según la ascendencia de la persona (se citó a Matsumoto et al. como referencia 9).
Se añadió texto para indicar que en un estudio que se realizó en los Países Bajos, las personas con BHD no presentaron una mayor prevalencia de cáncer colorrectal (CCR) en comparación con sus familiares sin esta afección, a pesar de que para las personas con BHD se usaron pruebas de detección del CCR ampliadas (se citó a Van de Beek et al. como referencia 29).
El Consejo editorial del PDQ sobre la genética del cáncer es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este resumen del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre la genética del síndrome de Birt-Hogg-Dubé. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El Consejo editorial del PDQ sobre la genética del cáncer, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
- Si el artículo se debe analizar en una reunión del consejo.
- Si conviene añadir texto acerca del artículo.
- Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Síndrome de Birt-Hogg-Dubé son:
- Alexandra Perez Lebensohn, MS, CGC (National Cancer Institute)
- Brian Matthew Shuch, MD (UCLA Health)
- Ramaprasad Srinivasan, MD, PhD (National Cancer Institute)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El Consejo editorial del PDQ sobre la genética del cáncer emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como “En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]”.
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
PDQ® sobre la genética del cáncer. PDQ Síndrome de Birt-Hogg-Dubé. Bethesda, MD: National Cancer Institute. Actualización:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
La información en estos resúmenes no se debe utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Comuníquese con el Instituto Nacional del Cáncer
Para obtener más información sobre las opciones para comunicarse con el NCI, incluso la dirección de correo electrónico, el número telefónico o el chat, consultar la página del Servicio de Información de Cáncer del Instituto Nacional del Cáncer.