Instituto Nacional del Cáncer
encontrar mi
Fecha de publicación: Jul 4, 2024
Información sobre el tratamiento de la histiocitosis de células de Langerhans (HCL): observación sola, cirugía, radioterapia o medicamentos orales, tópicos e intravenosos. El tratamiento se basa en el sitio y extensión de la enfermedad. Resumen para profesionales de la salud.
Tratamiento de la histiocitosis de células de Langerhans
Información general sobre la histiocitosis de células de Langerhans
Las enfermedades histiocíticas en niños y adultos se deben a la acumulación anómala de células del sistema mononuclear fagocítico. En este resumen solo se trata la histiocitosis de células de Langerhans (HCL), un trastorno de las células dendríticas de origen mieloide.
Las enfermedades histiocíticas se han reclasificado en 5 categorías; la HCL pertenece al grupo L (consultar el Cuadro 1). La HCL es consecuencia de la proliferación clonal de células de la HCL morfológicamente redondeadas e inmaduras desde el punto de vista inmunofenotípico y funcional, que se encuentran en las lesiones relevantes acompañadas de eosinófilos, macrófagos, linfocitos y, en ocasiones, células gigantes multinucleadas. Los histiocitos patológicos y las células de Langerhans (CL) normales de la epidermis comparten idénticas características inmunofenotípicas, como la presencia de gránulos de Birbeck identificados por microscopía electrónica. Hay claras diferencias morfológicas, fenotípicas y de expresión génica entre la variante celular patológica que se encuentra en las lesiones de la HCL (células de la HCL) y la variante normal de CL, de ahí el término células de la HCL.
| Grupo de histiocitosis | Enfermedades | ||
|---|---|---|---|
| XGA = xantogranuloma del adulto; HCB = histiocitiosis cefálica benigna; HEG = histiocitosis eruptiva generalizada; LHH = linfohistiocitosis hemofagocítica; XGJ = xantogranuloma juvenil; HCL = histiocitosis de células de Langerhans; RHM = reticulohistiocitosis multicéntrica; XGN = xantogranuloma necrobiótico; HNP = histiocitosis nodular progresiva; ERD = enfermedad de Rosai-Dorfman; RHS = reticulohistiocitoma solitario; XD = xantoma diseminado. | |||
| aAdaptación de Emile et al. | |||
| bReproducción autorizada de Blood, volumen 135, número 16, Carlos Rodríguez-Galindo, Carl E. Allen, Langerhans cell histiocytosis, páginas 1319–1331, derechos de autor 2020, autorizada por Elsevier. | |||
| Grupo L | Histiocitosis de células de Langerhans (HCL) | ||
| Histiocitosis de células indeterminadas (HCI) | |||
| Enfermedad de Erdheim-Chester (EEC) | |||
| Histiocitosis de células de Langerhans y Enfermedad de Erdheim-Chester mixtas | |||
| Grupo C | Histiocitosis cutáneas diferentes a HCL | ||
| Familia de granulomas xantomatosos: XGJ, XGA, RHS, HCB, HEG, HNP | |||
| Familia sin granulomas xantomatosos: ERD cutánea, XGN y otras | |||
| Histiocitosis cutáneas diferentes a HCL con componente sistémico mayor | |||
| Familia de granulomas xantomatosos: xantoma diseminado (XD) | |||
| Familia sin granulomas xantomatosos: reticulohistiocitosis multicéntrica (RHM) | |||
| Grupo R | Enfermedad de Rosai-Dorfman familiar | ||
| Enfermedad de Rosai-Dorfman esporádica | |||
| Enfermedad de Rosai-Dorfman clásica | |||
| Enfermedad de Rosai-Dorfman extraganglionar | |||
| Enfermedad de Rosai-Dorfman con neoplasia o enfermedad inmunitaria | |||
| Sin clasificación | |||
| Grupo M | Histiocitosis malignas primarias | ||
| Histiocitosis malignas secundarias | |||
| Grupo H | Linfohistiocitosis hemofagocítica primaria: afecciones hereditarias monogénicas que producen LHH | ||
| Linfohistiocitosis hemofagocítica secundaria (LHH no mendeliana) | |||
| Linfohistiocitosis hemofagocítica de origen desconocido o indeterminado | |||
Ahora se ha demostrado que las células de HCL, conocidas durante muchos años por originarse en una proliferación clonal, probablemente se derivan de un precursor mieloide cuya proliferación se relaciona de manera uniforme con la activación de la vía de señalización MAPK/ERK.
Desde el punto de vista clínico, la HCL es una enfermedad heterogénea que a veces compromete un solo órgano (HCL monosistémica), el cual a su vez puede afectar un solo sitio (unifocal) o múltiples sitios (multifocal). Otras veces, afecta múltiples órganos (HCL multisistémica). La HCL multisistémica puede comprometer un número limitado de órganos o puede ser diseminada. El compromiso de órganos específicos como el hígado, el bazo y el sistema hematopoyético permite clasificar la HCL multisistémica en dos grupos según el riesgo de muerte por la enfermedad: riesgo alto (es decir, con compromiso multisistémico de órganos de riesgo) y riesgo bajo (es decir, sin compromiso multisistémico de órganos de riesgo).
References
- Rodriguez-Galindo C, Allen CE: Langerhans cell histiocytosis. Blood 135 (16): 1319-1331, 2020.
- Emile JF, Abla O, Fraitag S, et al.: Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 127 (22): 2672-81, 2016.
- Berres ML, Lim KP, Peters T, et al.: BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 211 (4): 669-83, 2014.
- Allen CE, Merad M, McClain KL: Langerhans-Cell Histiocytosis. N Engl J Med 379 (9): 856-868, 2018.
- Willman CL, Busque L, Griffith BB, et al.: Langerhans'-cell histiocytosis (histiocytosis X)--a clonal proliferative disease. N Engl J Med 331 (3): 154-60, 1994.
- Yu RC, Chu C, Buluwela L, et al.: Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet 343 (8900): 767-8, 1994.
Características histopatológicas, inmunológicas y citogenéticas de la histiocitosis de células de Langerhans
Célula de origen y correlatos biológicos
El histiocito patológico o la célula de la histiocitosis de células de Langerhans (HCL) tiene un perfil de expresión génica muy similar al de una célula dendrítica mieloide. En estudios también se demostró que la variante BRAF V600E se puede identificar en las células mononucleares de la sangre periférica y en el DNA extracelular circulante, por lo general, en pacientes con enfermedad diseminada. Esto indica que la HCL multisistémica surge de una variante somática en la médula ósea o de una célula precursora circulante, mientras que la enfermedad localizada surge de una variante que ocurre en una célula precursora en un sitio local.
La clasificación moderna de las enfermedades histiocíticas las subdivide en enfermedades de las células dendríticas, enfermedades de los monocitos y macrófagos, y neoplasias malignas verdaderas. La HCL es una enfermedad de las células dendríticas. El análisis exhaustivo de datos de matriz de expresión génica de las células de la HCL concuerda con el concepto de que la célula de Langerhans (CL) de la piel no es la célula de origen de la HCL. Más bien, es probable que el origen sea una célula progenitora hematopoyética antes de convertirse en una célula dendrítica mieloide dedicada que expresa los mismos antígenos (CD1a y CD207) que la CL cutánea. Este concepto se apoyó además en los informes que indicaban que el perfil de transcripción de las células de la HCL era distinto al de las células dendríticas mieloides y plasmocitoides, así como al perfil de las CL epidérmicas.
Ahora, la HCL se considera una neoplasia mieloide. Sin embargo, hay controversia sobre la naturaleza de esta enfermedad como neoplasia maligna verdadera o como neoplasia con un comportamiento clínico variable. La misma variante BRAF V600E se ha encontrado en muchos cánceres; no obstante, el gen BRAF con la alteración en V600E también se encuentra en nevos benignos, lo que posiblemente indica que la transformación maligna requiere otras variantes adicionales. Estos hallazgos han planteado la posibilidad de un tratamiento con terapias dirigidas. Hay varios ensayos en curso sobre el uso de inhibidores de BRAF y MEK para el tratamiento de la HCL en adultos y niños.
Para obtener más información, consultar las secciones Estudios citogenéticos y genómicos y Análisis de citocinas.
Características histopatológicas
Las células de la histiocitosis de Langerhans (células de la HCL) que se encuentran en las lesiones propias de la HCL son células dendríticas inmaduras, que conforman menos del 10 % de las células presentes en la lesión. Por lo habitual, son células ovaladas grandes, con abundante citoplasma rosado y un núcleo en forma de frijol cuando se usa la tinción de hematoxilina y eosina. Las células de la HCL se tiñen con anticuerpos anti-S100, anti-CD1a y anti-langerina (CD207). La tinción anti-CD1a o anti-langerina confirma el diagnóstico de HCL, pero se necesita cautela al establecer una correlación de este hallazgo con el cuadro clínico inicial en los órganos donde hay células CL normales.
Debido a que las células de la HCL activan otras células inmunitarias, las lesiones de la HCL también contienen histiocitos, linfocitos, macrófagos, neutrófilos, eosinófilos y fibroblastos, incluso a veces contienen células gigantes multinucleadas.
Se han descrito los siguientes tres hallazgos histopatológicos en el encéfalo relacionados con la HCL:
- Lesiones expansivas en las meninges o el plexo coroideo con células de la HCL positivas para CD1a y predominio de linfocitos positivos para CD8.
- Lesiones expansivas en espacios delimitados por tejido conjuntivo con células de la HCL positivas para CD1a y predominio de linfocitos positivos para CD8 que causan una respuesta inflamatoria y pérdida neuronal.
- Lesiones neurodegenerativas: contienen células que se tiñen con la proteína BRAF alterada y que son positivas para CD14, CD33 y CD163, lo que las caracteriza como células hematopoyéticas mieloides o monocíticas. Estas son las CL patológicas que migraron al encéfalo, no se tiñen con CD1a ni CD207, se transformaron y se parecen a la microglía.
Anomalías inmunitarias
Por lo general, la CL es la presentadora primaria de antígenos a los linfocitos T vírgenes. Sin embargo, en la HCL, la célula dendrítica patológica no produce una estimulación eficiente de las respuestas primarias de los linfocitos T. En la tinción de anticuerpos para los marcadores de células dendríticas, como los antígenos CD80, CD86 y de clase II, se observó que en la HCL las células anormales son células dendríticas inmaduras. Estas células son deficientes para la presentación de antígenos y proliferan a una tasa baja.
Se ha notificado aumento de células T reguladoras en pacientes con HCL. Se encontró que la población de células CD4 positivas, CD25 (alta) y FoxP3 (alta) comprende un 20 % de las células T y, en las lesiones, están en contacto con células de la HCL. Estas células T se encontraron en concentraciones más altas en la sangre periférica de los pacientes con HCL que en los controles y volvieron a una concentración normal cuando los pacientes estaban en remisión. Se han encontrado células T con funcionamiento deficiente que expresan receptores inhibidores PD-1, TIM3 y LAG-3 en lesiones de HCL, pero no en la sangre periférica de los pacientes. Las células T disfuncionales se acumulan en las lesiones de la HCL porque el PD-1 en la superficie celular se enlaza con el PD-L1 en las células dendríticas patológicas.
Estudios citogenéticos y genómicos
Características genómicas de la histiocitosis de células de Langerhans
Variantes de BRAF, NRAS y ARAF
El fundamento genómico de la histiocitosis de células de Langerhans (HCL) avanzó gracias a un informe de 2010 sobre la detección de una variante activadora del oncogén BRAF (V600E) en 35 de 61 casos (57 %). En múltiples informes posteriores se confirmó la presencia de variantes BRAF V600E en el 50 % o más de los casos de HCL en niños. También se han descrito otras variantes de BRAF que producen activación de la señalización. Las variantes de ARAF son infrecuentes en la HCL, pero cuando están presentes, también llevan a la activación de la vía RAS-MAPK.
En una serie de 100 pacientes, se estudió la variante BRAF V600E en sangre y médula ósea y se obtuvo un resultado positivo para la variante BRAF V600E en el 65 % de los pacientes cuando se usó un método de reacción en cadena de la polimerasa cuantitativa sensible. Las células circulantes con la variante BRAF V600E se pudieron detectar en todos los pacientes de riesgo alto y en un subgrupo de pacientes con enfermedad multisistémica de riesgo bajo. El alelo BRAF V600E se detectó en el ADN libre circulantes en el 100 % de los pacientes con HCL multisistémica con compromiso de órganos de riesgo, el 42 % de los pacientes con HCL sin compromiso de órganos de riesgo y el 14 % de los pacientes con HCL monosistémica.
En pacientes de riesgo alto, el hallazgo en la médula ósea de células madre positivas para CD34 que además exhibían la variante confirmó el origen de la HCL en las células dendríticas mieloides. En los pacientes con enfermedad de riesgo bajo, la variante se identificó en células dendríticas mieloides más maduras, lo que indica que el estadio del desarrollo celular en el momento en que aparece la variante somática es determinante para definir la extensión de la enfermedad en la HCL.
En un principio se notificó que la HCL pulmonar en adultos no era clonal en cerca del 75 % de los casos, pero al analizar las variantes de BRAF en un estudio posterior, se encontró que entre el 25 % al 50 % de los adultos con esta enfermedad presentaban variantes BRAF V600E. En otro estudio de 26 casos de HCL pulmonar se encontró que el 50 % tenía variantes BRAF V600E y el 40 % tenía variantes de NRAS. El número de variantes policlonales y monoclonales es aproximadamente el mismo. No se ha determinado si la clonalidad y las variantes en la vía del gen BRAF coinciden en los mismos pacientes, lo que podría indicar una afección reactiva en lugar de una afección neoplásica en la HCL con pulmón del fumador, y una neoplasia clonal en otros tipos de HCL.
En un estudio de 117 pacientes con HCL, 83 pacientes adultos con HCL pulmonar se sometieron a análisis molecular. Cerca del 90 % de estos pacientes tenía variantes en la vía MAPK.[Nivel de evidencia C3] De los 69 pacientes en quienes se analizaron las muestras de biopsia mediante un panel de secuenciación de última generación de 74 genes, el 36 % tenía variantes BRAF V600E, el 29 % tenía deleciones BRAF N486-P490, el 15 % tenía deleciones o variantes de MAP2K1 y el 4 % tenía variantes de NRAS. Solo un paciente tenía una variante de KRAS. Además, en 11 pacientes, las muestras de biopsia se analizaron mediante secuenciación del exoma completo. Se encontraron, en promedio, 14 variantes por paciente; esto es notablemente más alto que el promedio de 1 variante por paciente observada en el entorno pediátrico. No hubo correlaciones clínicas, como la presencia de una variante BRAF V600E y el consumo de cigarrillos. De los 117 pacientes con HCL, el 60 % presentó recaída.
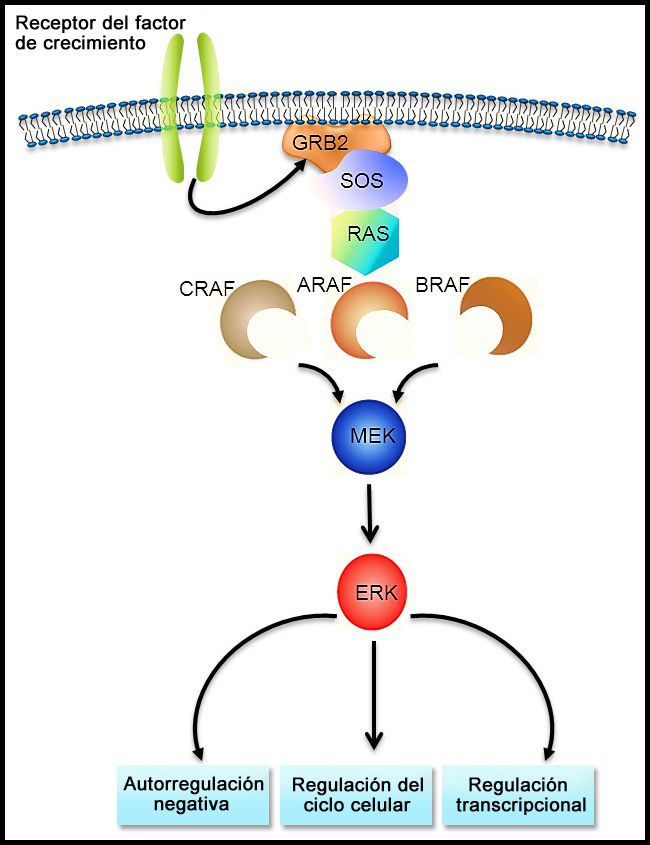 Figura 1. Cortesía de Rikhia Chakraborty, Ph.D. La autorización para reutilizar la figura en cualquier formato se debe solicitar directamente a la Dra. Chakraborty.
Figura 1. Cortesía de Rikhia Chakraborty, Ph.D. La autorización para reutilizar la figura en cualquier formato se debe solicitar directamente a la Dra. Chakraborty.
La vía de señalización RAS-MAPK (consultar la Figura 1) transmite señales desde un receptor de la superficie celular (por ejemplo, un factor de crecimiento) por la vía RAS (mediante una de las proteínas RAF [A, B o C]) de manera que se induce la fosforilación de MEK y después, de la cinasa regulada por señales extracelulares (ERK), lo que conduce a una señalización nuclear que afecta el ciclo celular y la regulación de la transcripción. La variante BRAF V600E produce fosforilación ininterrumpida y, por lo tanto, activación de MEK y ERK en ausencia de una señal externa. La activación de ERK ocurre por fosforilación, y esta ERK fosforilada se detecta en prácticamente todas las lesiones de HCL.
En un modelo murino de HCL, se observó que la presencia de la variante BRAF V600E inhibe la migración de las células dendríticas mediada por un receptor de quimiocina (CCR7), lo que las obliga a acumularse en la lesión de la HCL. Esta variante también causa un aumento de la expresión de BCL2L1, lo que produce resistencia a la apoptosis. Este proceso conlleva menor respuesta de las células a la quimioterapia. La variante BRAF V600E también interrumpe la proliferación de las células progenitoras hematopoyéticas y produce un fenotipo secretorio relacionado con la senescencia que promueve aún más la acumulación de células anormales.
Otro modelo murino con la variante BRAF V600E bajo el control de los promotores de los genes Scl o Map17 añadió conocimientos adicionales sobre las características biológicas de la HCL neurodegenerativa. En estos estudios se confirmó el origen hematopoyético de los macrófagos positivos para CD11a con variantes BRAF V600E. Este proceso rompe la barrera hematoencefálica y produce la pérdida de células de Purkinje, así como neurodegeneración progresiva mediante resistencia a la apoptosis y producción de proteínas secretoras relacionadas con la senescencia, que incluyen las citocinas inflamatorias IL-1, IL-6 y metaloproteinasas de matriz. El tratamiento con un inhibidor de la cinasa MAP y un senolítico (navitoclax) disminuyó el número de células patógenas y produjo mejoría clínica en los ratones.
En resumen, la HCL ahora se considera una neoplasia mieloide impulsada principalmente por variantes activadoras de la vía MAPK. Entre el 50 % y el 60 % de las variantes activadoras son variantes BRAF V600E, que abundan en pacientes con HCL multisistémica con compromiso de órganos de riesgo y en pacientes que tienen enfermedad neurodegenerativa. En los estudios en curso se evalúa si la detección de variantes de nivel bajo en la sangre periférica se puede usar como marcador de enfermedad residual mínima para ayudar en la toma de decisiones sobre el tratamiento.
Otras alteraciones en la vía RAS-MAPK
Debido a que es posible detectar la activación de la vía RAS-MAPK (elevación de ERK fosforilada) en todos los casos de HCL, incluso en aquellos sin variantes de BRAF, se consideró la posibilidad de que hubiera alteraciones genómicas en otros componentes de la vía. Se han identificado las siguientes alteraciones genómicas:
- Variantes de MAP2K1. La secuenciación del exoma completo en biopsias de tejido de HCL que exhiben alteraciones en el gen BRAF versus un gen BRAF natural reveló que 7 de 21 muestras con BRAF natural tenían variantes de MAP2K1; mientras que ninguna de las muestras con alteraciones en el gen BRAF tenía variantes de MAP2K1. Las variantes de MAP2K1 (que codifica MEK1) fueron activadoras, como se indica por la inducción de la fosforilación de ERK.
En otro estudio se detectaron variantes de MAP2K1 solo en 11 de 22 casos con gen BRAF natural. En un estudio se observó que la variante de MAP2K1 y otras variantes relacionadas con la HCL en niños y adultos son mutuamente excluyentes de la presencia de las variantes de BRAF. Los autores encontraron diversas variantes en otras vías (por ejemplo, JNK, RAS-ERK y JAK-STAT) en niños y adultos con la variante BRAF V600E o variantes de MAP2K1. En otro estudio se evaluaron alteraciones de cinasas y variantes mieloides en 73 adultos con HCL. Estos investigadores notificaron una mediana de 2 variantes por adulto, a diferencia de los niños que por lo general solo tienen 1 variante. Se encontró la variante BRAF V600E en el 31 % de los pacientes con HCL, una inserción y deleción (indel) en BRAF en el 29 % de ellos, y una variante de MAP2K1 en el 19 % de ellos. En el 89 % de los adultos con HCL se encontraron alteraciones en otras proteínas cinasas y vías relacionadas. Las variantes de MAP2K1 se presentaron en ausencia de variantes de BRAF.
- Deleciones en el marco de lectura de BRAF y fusiones génicas FAM73A::BRAF. Se han encontrado deleciones en el marco de lectura del gen BRAF y fusiones génicas en el marco de lectura de FAM73A::BRAF en el grupo de casos que no tienen variantes BRAF V600E ni de MAP2K1.
En resumen, los estudios corroboran la activación generalizada de ERK en la HCL, La activación de ERK en la mayoría de los casos se explica a partir de las alteraciones de los genes BRAF y MAP2K1. En conjunto, estas variantes en la vía de la cinasa MAP representan casi el 80 % de las causas de la activación generalizada de ERK en la HCL. El resto de los casos exhiben diversas variantes que incluyen deleciones pequeñas en BRAF, fusiones génicas de BRAF (mencionadas antes), así como variantes de los genes ARAF, MAP3K1, NRAS, ERBB3, PI3CA y CSF1R, así como otros genes infrecuentes. [Nivel de evidencia C1]
Repercusiones clínicas
Las repercusiones clínicas de los hallazgos genómicos descritos son las siguientes:
- La HCL se agrupa con otros tumores del ámbito pediátrico que tienen variantes activadoras de BRAF, entre ellos, algunas afecciones benignas (por ejemplo, nevos benignos) y neoplasias malignas de grado bajo (por ejemplo, astrocitoma pilocítico). Por lo general, todas estas afecciones tienen una evolución poco activa y en algunos casos se resuelven de manera espontánea. Esta evolución clínica característica quizás sea una manifestación de senescencia causada por oncogenes.
- En algunos estudios pediátricos, las variantes BRAF V600E se relacionaron con enfermedad multisistémica más grave, fracaso del tratamiento, aumento de las reactivaciones y mayor riesgo de neurodegeneración (ver más adelante). Estas correlaciones clínicas se investigaron hace poco con el fin de detectar otras variantes diferentes a la variante BRAF V600E. De manera similar a la cohorte con variantes BRAF V600E, todos los pacientes con HCL multisistémica y compromiso de órganos de riesgo exhibían variantes detectables en las células mononucleares de sangre periférica. De 7 pacientes con HCL multisistémica sin compromiso de órganos de riesgo, 4 tenían variantes detectables. Ningún paciente con enfermedad monosistémica presentó variantes detectables en las células mononucleares de sangre periférica. Los autores concluyeron que otras variantes en la vía MAPK se relacionan con el estado de riesgo, de manera similar a como lo hacen las variantes BRAF V600E.
Las variantes BRAF V600E son la diana terapéutica de los inhibidores de BRAF (por ejemplo, vemurafenib y dabrafenib) y de la combinación de inhibidores de BRAF con inhibidores de MEK (por ejemplo, dabrafenib y trametinib, o vemurafenib y cobimetinib). Estos fármacos y sus combinaciones están aprobadas para su uso en adultos con melanoma. En adultos, el tratamiento del melanoma con combinaciones de un inhibidor de BRAF y un inhibidor de MEK mostró una mejora significativa de los desenlaces de supervivencia sin progresión en comparación con el tratamiento con un inhibidor de BRAF solo.
En varios informes de casos y en 2 series de casos también se observó eficacia de los inhibidores de BRAF para el tratamiento de la HCL en niños. No obstante, es difícil evaluar la función a largo plazo de este tratamiento porque la mayoría de los pacientes recaen al suspender los inhibidores. Para obtener más información, consultar las secciones Tratamiento de la histiocitosis de células de Langerhans multisistémica recidivante, resistente al tratamiento o progresiva de riesgo alto y Terapias dirigidas para el tratamiento de la enfermedad monosistémica y multisistémica.
- Se encontraron células circulantes con variantes BRAF V600E en el 59 % de los pacientes con HCL y enfermedad neurodegenerativa en comparación con el 15 % de los pacientes con HCL sin enfermedad neurodegenerativa La detección de células circulantes que exhibe la variante tiene una sensibilidad de 0,59 y una especificidad de 0,86 para el desarrollo de enfermedad neurodegenerativa. Incluso después del tratamiento, algunos pacientes con HCL y enfermedad neurodegenerativa tienen células circulantes con variantes BRAF V600E.
- Es posible que nuevas investigaciones permitan usar la detección de la variante BRAF V600E (o quizás las variantes de MAP2K1) en células circulantes o en el DNA extracelular circulante como una herramienta diagnóstica útil para diferenciar la enfermedad de riesgo alto de la enfermedad de riesgo bajo. Además, para los pacientes que tienen una variante somática, la persistencia de las células circulantes con la variante puede ser útil como marcador de enfermedad residual.
Análisis de citocinas
Las pruebas con tinción inmunohistoquímica han mostrado un aumento de las concentraciones de muchas citocinas o quimiocinas diferentes, tanto en lesiones de HCL como en el suero o plasma de pacientes con HCL. En un análisis de la expresión génica en la HCL mediante técnicas de micromatriz génica, se identificaron 2000 genes expresados de manera diferencial. De 65 genes de los que se notificó antes que estaban relacionados con la HCL, solo 11 exhibieron expresión aumentada en los resultados de la micromatriz. El gen con mayor aumento de expresión en las células positivas para CD207 y CD3 fue el SPP1 (codificador de la proteína osteopontina); otros genes que activan y reclutan células T hacia los sitios de inflamación también exhiben un aumento de expresión. El perfil de expresión de las células T fue el de un fenotipo de células T reguladoras activadas con aumento de la expresión de FOXP3, CTLA4 y SPP1. Estos hallazgos respaldan un informe previo sobre el aumento de las células T reguladoras en la HCL. Hubo una expresión pronunciada de los genes relacionados con los progenitores mieloides tempranos, como CD33 y CD44, lo que es compatible con un informe anterior de aumento de las células dendríticas mieloides en la sangre de pacientes con HCL. Se planteó un modelo de precursores de células dendríticas mieloides equivocados en el que se reclutan células precursoras de células dendríticas mieloides a los sitios de HCL por un mecanismo desconocido, y las células dendríticas, a su vez, reclutan linfocitos mediante la excreción de osteopontina, neuropilina-1 y vanina-1.
En un estudio se evaluaron los posibles biomarcadores de la HCL en el sistema nervioso central. En el estudio se examinaron 121 proteínas únicas en el líquido cefalorraquídeo (LCR) de 40 pacientes pediátricos con HCL que se compararon con un grupo de control que incluyó a 29 pacientes con leucemia linfoblástica aguda, 25 pacientes con tumores encefálicos, 28 pacientes con enfermedades neurodegenerativas y 9 pacientes con linfohistiocitosis hemofagocítica. Solo la osteopontina exhibió un aumento significativo de la concentración en el LCR de los pacientes de HCL con neurodegeneración o lesiones expansivas (hipófisis), en comparación con todos los grupos de control. El análisis de la expresión de osteopontina en estos tejidos confirmó aumento de la expresión del gen SPP1.
Varios investigadores han publicado estudios en los que analizan la concentración de diversas citocinas o factores de crecimiento en la sangre de pacientes con HCL. En estos estudios se incluyeron muchos de los genes cuya expresión no estaba aumentada según los resultados de expresión génica mencionados antes. Una explicación de las concentraciones elevadas de estas proteínas es una respuesta inflamatoria sistémica, donde las células que se encuentran fuera de las lesiones de la HCL producen citocinas y factores de crecimiento. Otra posible explicación es que los macrófagos de las lesiones de la HCL producen las citocinas que se cuantifican en la sangre o que están concentradas en las lesiones.
Se midieron las concentraciones de IL-1 ß y prostaglandina GE2 en la saliva de pacientes con lesiones orales de HCL o HCL multisistémica de riesgo alto con lesiones orales y sin estas. Las concentraciones de ambas sustancias estaban más elevadas en los pacientes con enfermedad activa y disminuyeron después de un tratamiento exitoso.
References
- Allen CE, Li L, Peters TL, et al.: Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol 184 (8): 4557-67, 2010.
- Berres ML, Lim KP, Peters T, et al.: BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 211 (4): 669-83, 2014.
- Hyman DM, Diamond EL, Vibat CR, et al.: Prospective blinded study of BRAFV600E mutation detection in cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov 5 (1): 64-71, 2015.
- Emile JF, Abla O, Fraitag S, et al.: Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 127 (22): 2672-81, 2016.
- Picarsic J, Jaffe R: Nosology and Pathology of Langerhans Cell Histiocytosis. Hematol Oncol Clin North Am 29 (5): 799-823, 2015.
- Ginhoux F, Merad M: Ontogeny and homeostasis of Langerhans cells. Immunol Cell Biol 88 (4): 387-92, 2010 May-Jun.
- Durham BH, Roos-Weil D, Baillou C, et al.: Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood 130 (2): 176-180, 2017.
- Hutter C, Kauer M, Simonitsch-Klupp I, et al.: Notch is active in Langerhans cell histiocytosis and confers pathognomonic features on dendritic cells. Blood 120 (26): 5199-208, 2012.
- Berres ML, Allen CE, Merad M: Pathological consequence of misguided dendritic cell differentiation in histiocytic diseases. Adv Immunol 120: 127-61, 2013.
- Badalian-Very G, Vergilio JA, Fleming M, et al.: Pathogenesis of Langerhans cell histiocytosis. Annu Rev Pathol 8: 1-20, 2013.
- Geissmann F, Lepelletier Y, Fraitag S, et al.: Differentiation of Langerhans cells in Langerhans cell histiocytosis. Blood 97 (5): 1241-8, 2001.
- Chikwava K, Jaffe R: Langerin (CD207) staining in normal pediatric tissues, reactive lymph nodes, and childhood histiocytic disorders. Pediatr Dev Pathol 7 (6): 607-14, 2004 Nov-Dec.
- McClain KL, Picarsic J, Chakraborty R, et al.: CNS Langerhans cell histiocytosis: Common hematopoietic origin for LCH-associated neurodegeneration and mass lesions. Cancer 124 (12): 2607-2620, 2018.
- Yu RC, Morris JF, Pritchard J, et al.: Defective alloantigen-presenting capacity of 'Langerhans cell histiocytosis cells'. Arch Dis Child 67 (11): 1370-2, 1992.
- Senechal B, Elain G, Jeziorski E, et al.: Expansion of regulatory T cells in patients with Langerhans cell histiocytosis. PLoS Med 4 (8): e253, 2007.
- Sengal A, Velazquez J, Hahne M, et al.: Overcoming T-cell exhaustion in LCH: PD-1 blockade and targeted MAPK inhibition are synergistic in a mouse model of LCH. Blood 137 (13): 1777-1791, 2021.
- Badalian-Very G, Vergilio JA, Degar BA, et al.: Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 116 (11): 1919-23, 2010.
- Satoh T, Smith A, Sarde A, et al.: B-RAF mutant alleles associated with Langerhans cell histiocytosis, a granulomatous pediatric disease. PLoS One 7 (4): e33891, 2012.
- Sahm F, Capper D, Preusser M, et al.: BRAFV600E mutant protein is expressed in cells of variable maturation in Langerhans cell histiocytosis. Blood 120 (12): e28-34, 2012.
- Héritier S, Hélias-Rodzewicz Z, Chakraborty R, et al.: New somatic BRAF splicing mutation in Langerhans cell histiocytosis. Mol Cancer 16 (1): 115, 2017.
- Nelson DS, Quispel W, Badalian-Very G, et al.: Somatic activating ARAF mutations in Langerhans cell histiocytosis. Blood 123 (20): 3152-5, 2014.
- Héritier S, Hélias-Rodzewicz Z, Lapillonne H, et al.: Circulating cell-free BRAF(V600E) as a biomarker in children with Langerhans cell histiocytosis. Br J Haematol 178 (3): 457-467, 2017.
- Dacic S, Trusky C, Bakker A, et al.: Genotypic analysis of pulmonary Langerhans cell histiocytosis. Hum Pathol 34 (12): 1345-9, 2003.
- Roden AC, Hu X, Kip S, et al.: BRAF V600E expression in Langerhans cell histiocytosis: clinical and immunohistochemical study on 25 pulmonary and 54 extrapulmonary cases. Am J Surg Pathol 38 (4): 548-51, 2014.
- Mourah S, How-Kit A, Meignin V, et al.: Recurrent NRAS mutations in pulmonary Langerhans cell histiocytosis. Eur Respir J 47 (6): 1785-96, 2016.
- Jouenne F, Chevret S, Bugnet E, et al.: Genetic landscape of adult Langerhans cell histiocytosis with lung involvement. Eur Respir J 55 (2): , 2020.
- Chakraborty R, Burke TM, Hampton OA, et al.: Alternative genetic mechanisms of BRAF activation in Langerhans cell histiocytosis. Blood 128 (21): 2533-2537, 2016.
- Chakraborty R, Hampton OA, Shen X, et al.: Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood 124 (19): 3007-15, 2014.
- Hogstad B, Berres ML, Chakraborty R, et al.: RAF/MEK/extracellular signal-related kinase pathway suppresses dendritic cell migration and traps dendritic cells in Langerhans cell histiocytosis lesions. J Exp Med 215 (1): 319-336, 2018.
- Bigenwald C, Le Berichel J, Wilk CM, et al.: BRAFV600E-induced senescence drives Langerhans cell histiocytosis pathophysiology. Nat Med 27 (5): 851-861, 2021.
- Wilk CM, Cathomas F, Török O, et al.: Circulating senescent myeloid cells infiltrate the brain and cause neurodegeneration in histiocytic disorders. Immunity 56 (12): 2790-2802.e6, 2023.
- Milne P, Abhyankar H, Scull B, et al.: Cellular distribution of mutations and association with disease risk in Langerhans cell histiocytosis without BRAFV600E. Blood Adv 6 (16): 4901-4904, 2022.
- Brown NA, Furtado LV, Betz BL, et al.: High prevalence of somatic MAP2K1 mutations in BRAF V600E-negative Langerhans cell histiocytosis. Blood 124 (10): 1655-8, 2014.
- Durham BH, Lopez Rodrigo E, Picarsic J, et al.: Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med 25 (12): 1839-1842, 2019.
- Chen J, Zhao AL, Duan MH, et al.: Diverse kinase alterations and myeloid-associated mutations in adult histiocytosis. Leukemia 36 (2): 573-576, 2022.
- Michaloglou C, Vredeveld LC, Soengas MS, et al.: BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436 (7051): 720-4, 2005.
- Jones DT, Kocialkowski S, Liu L, et al.: Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68 (21): 8673-7, 2008.
- Pfister S, Janzarik WG, Remke M, et al.: BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 118 (5): 1739-49, 2008.
- Jacob K, Quang-Khuong DA, Jones DT, et al.: Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res 17 (14): 4650-60, 2011.
- Héritier S, Emile JF, Barkaoui MA, et al.: BRAF Mutation Correlates With High-Risk Langerhans Cell Histiocytosis and Increased Resistance to First-Line Therapy. J Clin Oncol 34 (25): 3023-30, 2016.
- Larkin J, Ascierto PA, Dréno B, et al.: Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 371 (20): 1867-76, 2014.
- Long GV, Stroyakovskiy D, Gogas H, et al.: Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 386 (9992): 444-51, 2015.
- Eckstein OS, Visser J, Rodriguez-Galindo C, et al.: Clinical responses and persistent BRAF V600E+ blood cells in children with LCH treated with MAPK pathway inhibition. Blood 133 (15): 1691-1694, 2019.
- Donadieu J, Larabi IA, Tardieu M, et al.: Vemurafenib for Refractory Multisystem Langerhans Cell Histiocytosis in Children: An International Observational Study. J Clin Oncol 37 (31): 2857-2865, 2019.
- Kolenová A, Schwentner R, Jug G, et al.: Targeted inhibition of the MAPK pathway: emerging salvage option for progressive life-threatening multisystem LCH. Blood Adv 1 (6): 352-356, 2017.
- Lee LH, Gasilina A, Roychoudhury J, et al.: Real-time genomic profiling of histiocytoses identifies early-kinase domain BRAF alterations while improving treatment outcomes. JCI Insight 2 (3): e89473, 2017.
- Héritier S, Jehanne M, Leverger G, et al.: Vemurafenib Use in an Infant for High-Risk Langerhans Cell Histiocytosis. JAMA Oncol 1 (6): 836-8, 2015.
- Váradi Z, Bánusz R, Csomor J, et al.: Effective BRAF inhibitor vemurafenib therapy in a 2-year-old patient with sequentially diagnosed Langerhans cell histiocytosis and Erdheim-Chester disease. Onco Targets Ther 10: 521-526, 2017.
- Fleming MD, Pinkus JL, Fournier MV, et al.: Coincident expression of the chemokine receptors CCR6 and CCR7 by pathologic Langerhans cells in Langerhans cell histiocytosis. Blood 101 (7): 2473-5, 2003.
- Annels NE, Da Costa CE, Prins FA, et al.: Aberrant chemokine receptor expression and chemokine production by Langerhans cells underlies the pathogenesis of Langerhans cell histiocytosis. J Exp Med 197 (10): 1385-90, 2003.
- Rolland A, Guyon L, Gill M, et al.: Increased blood myeloid dendritic cells and dendritic cell-poietins in Langerhans cell histiocytosis. J Immunol 174 (5): 3067-71, 2005.
- Preliasco VF, Benchuya C, Pavan V, et al.: IL-1 beta and PGE2 levels are increased in the saliva of children with Langerhans cell histiocytosis. J Oral Pathol Med 37 (9): 522-7, 2008.
Histiocitosis de células de Langerhans infantil
Información general sobre la histiocitosis de células de Langerhans infantil
Incidencia
Se calcula que la incidencia anual de histiocitosis de células de Langerhans (HCL) oscila entre 2 y 10 casos por 1 millón de personas de 15 años o menos. La proporción entre sexos (H:M) es cercana a 1, y la mediana de la edad en el momento del cuadro clínico inicial es de 30 meses. En una encuesta de 4 años realizada en Francia a 251 casos nuevos de HCL, se encontró una incidencia anual de 4,6 casos por 1 millón de personas menores de 15 años (H:M, 1,2).
En un estudio poblacional en Inglaterra se identificaron 658 pacientes que recibieron el diagnóstico de HCL entre 2013 y 2019. La prevalencia de HCL fue de 9,95 casos por 1 millón de personas a finales de 2019. Entre ellos, el 49 % de los pacientes eran menores de 15 años, con una tasa de incidencia de 4,46 casos por 1 millón de niños por año. Los autores consideraron que esta incidencia probablemente sea una subestimación, en especial para la HCL monosistémica. Este es el primer estudio que en el que se identifica con exactitud pacientes adultos de 30 a 60 años o más. Sin embargo, el estudio también incluyó a pacientes de 15 a 29 años en la categoría de adultos, lo que produjo una tasa de incidencia total en adultos de 1,06 casos por 1 millón de adultos por año. Los pacientes que vivían en circunstancias socioeconómicas más desfavorecidas y los mayores de 30 años tuvieron tasas de supervivencia más precarias que las personas con un nivel socioeconómico más alto o que los niños.
Se revisaron los datos del registro del Surveillance, Epidemiology, and End Results (SEER) de 2000 a 2009 para identificar los casos de HCL de riesgo alto y evaluar las variables demográficas. De 145 casos, la incidencia estandarizada por edad de la enfermedad diseminada fue de 0,7 por 1 millón de niños por año, con incidencia más baja en los pacientes negros (0,41 por 1 millón) y más alta en los pacientes hispanos (1,63 por 1 millón) menores de 5 años. El hacinamiento y unas condiciones socioeconómicas más precarias fueron factores relacionados con mayor riesgo de HCL, tal vez por la correlación con infecciones maternas y neonatales. En un estudio poblacional de casos y controles, las madres hispanas tuvieron más probabilidad que las madres blancas no hispanas de tener hijos con HCL; este riesgo aumentó cuando ambos progenitores eran hispanos. Las madres negras no hispanas tuvieron menos probabilidades que las madres blancas no hispanas de tener niños con HCL. Además, en un estudio familiar de asociación genómica, se encontró una asociación fuerte entre un polimorfismo del gen SMAD6 y la HCL, en especial en pacientes hispanos. El estudio de Inglaterra (descrito antes) incluyó a 658 adultos y niños, el 79 % de los cuales eran blancos. En este estudio no se observó ningún aumento de la incidencia en la población hispana, lo que refleja las diferencias en la población de Inglaterra.
Factores de riesgo
Aunque se han propuesto los siguientes factores de riesgo de la HCL, no hay confirmación de asociaciones firmes y constantes:
- Exposición parental a disolventes.
- Antecedentes familiares de cáncer.
- Antecedentes personales o familiares de enfermedad tiroidea.
- Infecciones perinatales.
- Exposición ocupacional de los progenitores al polvo de metal y granito o al aserrín.
- Origen étnico y raza hispanos.
- Nivel socioeconómico bajo.
- Vacunas incompletas durante la niñez.
Los esfuerzos para definir una causa viral no han tenido éxito.
Evaluación diagnóstica
La evaluación completa de cualquier paciente que presente HCL incluye lo siguiente:
- Anamnesis y examen físico: evaluación completa con atención especial a la piel, los ganglios linfáticos, los oídos, la orofaringe, las encías, la lengua, los dientes, los huesos, los pulmones, la tiroides, el tamaño del hígado y el bazo, las anomalías óseas, la velocidad de crecimiento, y los antecedentes de polidipsia y polaquiuria.
Otras pruebas y procedimientos son los siguientes:
- Análisis de sangre: abarcan un recuento sanguíneo completo con diferencial de leucocitos y plaquetas, pruebas de funcionamiento hepático (por ejemplo, bilirrubina, albúmina, aspartato–aminotransferasa, alanina–aminotransferasa, γ-glutamil–transferasa, tiempo de protrombina, tiempo parcial de tromboplastina o índice internacional estandarizado [INR] en pacientes con hepatomegalia, ictericia, enzimas hepáticas elevadas o albúmina baja) y electrólitos en suero.
En pacientes con HCL multisistémica grave, es posible que sea necesario obtener otras pruebas para una linfohistiocitosis hemofagocítica secundaria, como ferritina, triglicéridos, fibrinógeno, dímeros d, lactato–deshidrogenasa, CXCL9 y sCD25.
- Evaluación de la vía RAS-RAF-MEK: aunque la evaluación de la vía RAS-RAF-MEK no se exige como parte del proceso diagnóstico para los pacientes con HCL, es posible detectar la variante de BRAF mediante pruebas inmunohistoquímicas o métodos de diagnóstico molecular en tejido fresco y fijado con formol y sangre periférica.
- Análisis de orina: abarcan el urianálisis y una prueba de la sed si se sospecha diabetes insípida. Las pruebas de la sed en niños muy pequeños, en especial, en lactantes, se realizan bajo observación médica estricta.
- Aspiración de médula ósea con biopsia: se indica para pacientes con enfermedad multisistémica que tienen anemia o trombocitopenia inexplicadas. Las muestras de la biopsia se deben teñir con inmunotinción anti-CD1a o anti-CD207 (langerina) y con inmunotinción anti-CD163 para facilitar la detección de las células de la HCL. También es importante obtener un análisis de reacción en cadena de la polimerasa (PCR) para detectar las células con alteraciones en BRAF.
- Pruebas radiológicas y con imágenes: para el primer nivel de detección se usan series óseas y craneales, gammagrafías óseas y radiografía del tórax. Las tomografías por emisión de positrones (TEP) se utilizan cada vez más debido a su superioridad para el diagnóstico y la evaluación de la respuesta al tratamiento, en comparación con las gammagrafías óseas.
- Tomografía computarizada (TC): en ocasiones se indica una TC de la cabeza si se sospecha compromiso de la órbita, la apófisis mastoides o el área maxilofacial. Las pruebas por imágenes pueden incluir imágenes por resonancia magnética (IRM) con contraste de gadolinio del encéfalo para pacientes con diabetes insípida o sospecha de compromiso encefálico o vertebral.
La TC pulmonar a veces se indica en pacientes con radiografías del tórax anormales o síntomas pulmonares. Es posible que las TC de alta resolución muestren indicios de HCL pulmonar cuando la radiografía de tórax es normal. Por lo tanto, se puede considerar una TC cuando hay signos o síntomas respiratorios en lactantes y niños pequeños que tienen radiografías de tórax normales. Los pacientes con HCL pulmonar también pueden tener radiografías de tórax normales y pruebas del funcionamiento pulmonar anormales.
La HCL causa cambios de hígado graso o áreas hipodensas a lo largo del espacio porta, que se pueden identificar mediante TC, si está indicada.
- TEP con flúor F 18-fludesoxiglucosa (18F-FDG): se notificaron anomalías en la TEP con 18F-FDG del encéfalo en 7 pacientes con HCL que exhibían signos neurológicos y radiográficos de enfermedad neurodegenerativa. La correlación entre los hallazgos de la IRM en la sustancia blanca cerebelosa fue buena, pero lo fue menos en los núcleos caudados y la corteza frontal. Se ha indicado que la TEP permite detectar antes que la IRM las anomalías de los pacientes en riesgo alto de HCL neurodegenerativa. Las TEP a menudo muestran lesiones que no se encuentran con otros métodos y muestran también la disminución de la actividad de la HCL después de 6 semanas de tratamiento, lo que proporciona una mejor evaluación de la respuesta al tratamiento que las gammagrafías óseas o las radiografías simples. Sin embargo, en un estudio se indica que las gammagrafías óseas son más sensibles que las TEP para detectar lesiones en las manos y los pies.
- Imágenes por resonancia magnética (IRM): los hallazgos de una IRM en pacientes con diabetes insípida incluyen engrosamiento y nodularidad del infundíbulo hipofisario con pérdida del punto brillante de la hipófisis posterior, lo que refleja la ausencia de la hormona antidiurética.
Todos los pacientes con compromiso de cuerpo vertebral necesitan una evaluación detenida del tejido blando circundante que a veces afecta la médula espinal.
Los hallazgos de la IRM compatibles con una HCL del sistema nervioso central (SNC) incluyen la intensificación de la protuberancia, los ganglios basales y la sustancia blanca cerebelosa cuando se usa la técnica FLAIR en T2, además de lesiones expansivas o realce meníngeo. En un informe de 163 pacientes, se encontraron lesiones meníngeas en un 29 % de los pacientes y compromiso del plexo coroideo en un 6 %. Se encontraron lesiones en los senos paranasales o lesiones mastoideas en el 55 % de los pacientes versus el 20 % de los controles, y se encontraron espacios de Virchow-Robin acentuados en el 70 % de los pacientes versus el 27 % de los controles.
- Tomografía computarizada (TC): en ocasiones se indica una TC de la cabeza si se sospecha compromiso de la órbita, la apófisis mastoides o el área maxilofacial. Las pruebas por imágenes pueden incluir imágenes por resonancia magnética (IRM) con contraste de gadolinio del encéfalo para pacientes con diabetes insípida o sospecha de compromiso encefálico o vertebral.
- Biopsia: las lesiones óseas líticas, la piel y los ganglios linfáticos son los sitios más frecuentes para obtener biopsias diagnósticas de la HCL. Se indica una biopsia del hígado cuando un niño con HCL presenta hipoalbuminemia que no es causada por una HCL gastrointestinal u otra causa. Estos pacientes, por lo habitual, tienen concentraciones elevadas de bilirrubina o de enzimas hepáticas. Cuando el lavado broncoalveolar no es diagnóstico, es posible que sea necesaria una biopsia abierta de pulmón con el fin de extraer tejido para el diagnóstico de una HCL pulmonar. El diagnóstico del compromiso gastrointestinal en la HCL es difícil por el compromiso irregular. En general, se necesita un examen endoscópico minucioso que incluya varias biopsias.
El diagnóstico definitivo siempre exige un diagnóstico patológico. No obstante, algunas veces es complicado obtener la confirmación patológica o hay una contraindicación para la biopsia, como una enfermedad aislada del infundíbulo hipofisario o una vértebra plana sin masa de tejido blando, y en este caso el riesgo supera el beneficio de obtener la confirmación del diagnóstico.
Factores pronósticos
El pronóstico esta muy relacionado con la extensión de la enfermedad en el momento del cuadro clínico inicial cuando hay compromiso de órganos de riesgo alto (hígado, bazo o médula ósea) y con la respuesta al tratamiento inicial. En muchos estudios se confirmó una tasa alta de mortalidad (35 %) en pacientes con enfermedad multisistémica de riesgo alto, cuando no hay buena respuesta al tratamiento durante las primeras 6 semanas. Debido a los avances del tratamiento, como la introducción temprana de terapia complementaria para los pacientes con una respuesta deficiente, el desenlace para los niños con HCL y compromiso de órganos de riesgo alto ha mejorado. En los datos de HISTSOC-LCH-III (NCT00276757) se observó una tasa de supervivencia general (SG) de un 84 % en los pacientes tratados durante 12 meses con quimioterapia sistémica.
Durante muchos años, se pensó que los pulmones eran órganos de riesgo alto, pero el compromiso pulmonar aislado en la HCL pediátrica ya no se considera que acarrea un riesgo significativo de muerte, a menos que se presente neumotórax unilateral o bilateral.
Los pacientes con enfermedad monosistémica y con enfermedad multisistémica de riesgo bajo no suelen morir a causa de la HCL, pero la enfermedad recidivante produce muchas complicaciones mórbidas y efectos tardíos significativos. En general, se encontraron recidivas en el 10 % de los pacientes con enfermedad unifocal monosistémica, el 25 % de los pacientes con HCL ósea multifocal monosistémica y el 50 % de los pacientes con enfermedad multisistémica de riesgo bajo, así como en aquellos con enfermedad multisistémica de riesgo alto que logran un estado de enfermedad inactiva con quimioterapia. En los datos del estudio HISTSOC-LCH-III se observó una diferencia significativa de la tasa de reactivación en los pacientes con compromiso de órganos de riesgo bajo asignados al azar para recibir 6 meses de tratamiento (54 %) versus 12 meses de tratamiento (37 %). De manera similar, un grupo de pacientes de riesgo alto que se trataron de manera no aleatorizada durante 12 meses tuvo una tasa de reactivación del 30 %, en comparación con más del 50 % en estudios previos donde los pacientes recibieron la misma terapia durante 6 meses.
En la mayoría de los pacientes de riesgo alto cuya enfermedad se reactiva (30 %) después de lograr un estado de enfermedad inactiva, esta reactivación se produce en órganos de riesgo bajo como el hueso. Estos pacientes tendrán el mismo riesgo de efectos tardíos que los pacientes con enfermedad multisistémica de riesgo bajo. El principal desafío del tratamiento actual es reducir esta incidencia general de reactivaciones en un 20 % a un 30 %, además del riesgo significativo de consecuencias permanentes graves en este grupo de pacientes.
Además del alcance de la enfermedad, los factores pronósticos para los niños con HCL son los siguientes:
- Edad en el momento del diagnóstico. Aunque antes se pensaba que una edad inferior a 2 años presagiaba un pronóstico precario, los datos del estudio HISTSOC-LCH-II indicaron que los pacientes de 2 años o menos sin compromiso de órganos de riesgo alto tuvieron la misma respuesta al tratamiento que los pacientes de más edad. Por el contrario, la SG fue más precaria en los neonatos con compromiso de órganos de riesgo, en comparación con los lactantes y los niños con la misma extensión de la enfermedad, cuando los pacientes recibieron tratamiento durante solo 6 meses.
- Respuesta al tratamiento. Se ha demostrado que la respuesta al tratamiento entre las 6 y 12 semanas es un factor pronóstico más importante que la edad. La duración e intensidad del tratamiento influyen en la respuesta general al mismo.
- Sitios de compromiso.
- Variantes de BRAF o MAP2K1.
En un estudio de 173 pacientes con la variante BRAF V600E y 142 pacientes sin esta variante, se identificó la variante en el 88 % de los pacientes con enfermedad de riesgo alto, el 69 % de los pacientes con HCL multisistémica de riesgo bajo y el 44 % de los pacientes con HCL monosistémica de riesgo bajo. La variante también se encontró en el 75 % de los pacientes con síndrome neurodegenerativo y en el 73 % de los pacientes con compromiso hipofisario. La variante BRAF V600E también se ha relacionado con un aumento de la incidencia de enfermedad cutánea y una edad más temprana de presentación inicial. La resistencia al tratamiento inicial y la recaída fueron más altas en los pacientes que tenían la variante. Las variantes de MAP2K1 se asociaron con enfermedad monosistémica ósea.
En un estudio anterior de 100 pacientes no se encontró ninguna de esas correlaciones clínicas, excepto que las recaídas se presentaron con mayor frecuencia en pacientes con HCL de riesgo bajo y riesgo alto que tenían la variante BRAF V600E.
En un estudio colaborativo internacional de 377 pacientes, se encontraron 300 pacientes (79,6 %) con variantes en la vía MAPK y se compararon con pacientes sin variantes. En este estudio se confirmaron los resultados de un estudio anterior. También se encontró un aumento del riesgo de HCL ósea de riesgo para el SNC y compromiso gastrointestinal y cutáneo, y menos casos de HCL ósea multifocal monosistémica con variantes de BRAF en pacientes con variantes en la vía MAPK. Una cohorte de pacientes con una variante del exón 12 en el gen BRAF tuvo una incidencia mucho más alta de HCL pulmonar. Las variantes de MAP2K1 fueron más frecuentes en pacientes con HCL ósea monosistémica, pero no en pacientes con HCL ósea de riesgo para el SNC. La repercusión pronóstica de la variante de BRAF se relacionó de manera más estrecha con el compromiso multisistémico y de órganos de riesgo, en lugar de la presencia de la variante en sí.
Una proporción significativa de pacientes que sobreviven a la HCL presentan recaídas de la enfermedad o consecuencias permanentes. La diabetes insípida de origen central es la afección más común y la HCL neurodegenerativa del SNC es la afección más grave.
Consideraciones para el seguimiento de la histiocitosis de células de Langerhans infantil
Los pacientes se deben someter a un seguimiento durante muchos años, debido al riesgo de reactivación (que oscila entre un 10 % para las lesiones óseas monosistémicas unifocales hasta casi un 50 % para la HCL multisistémica de riesgo bajo y alto) y el riesgo de efectos permanentes a largo plazo.
Los pacientes con diabetes insípida o lesiones craneales en los huesos de la órbita, la apófisis mastoides o el temporal exhiben un riesgo más alto de una HCL con compromiso del sistema nervioso central (SNC) y una HLC en el SNC con síndrome neurodegenerativo. Estos pacientes se deben someter a IRM con contraste de gadolinio en el momento del diagnóstico de la HCL y, luego, cada 1 o 2 años durante 10 años para detectar indicios de enfermedad en el SNC. El comité sobre la HCL en el SNC de la Histiocyte Society no recomienda ningún tratamiento cuando se encuentran indicios radiológicos de HCL en el SNC de tipo neurodegenerativo en ausencia de neurodegeneración clínica y los hallazgos de la IRM no cambian con el tiempo. Sin embargo, se recomiendan exámenes neurológicos cuidadosos e imágenes apropiadas de IRM a intervalos regulares.
También se deben obtener pruebas de respuesta auditiva del tronco encefálico a intervalos regulares para identificar lo antes posible las manifestaciones clínicas de la HCL del SNC, ya que esto quizás afecte la respuesta al tratamiento. Cuando hay signos clínicos, se indica la intervención en pacientes con evidencia radiológica de cambios en el cerebelo relacionados con la HCL. En los estudios disponibles de las diferentes formas de tratamiento para la neurodegeneración del SNC, se indica que a veces los cambios neurodegenerativos se estabilizan o mejoran, pero solo con un tratamiento precoz. Es fundamental vigilar a los pacientes en riesgo con exámenes neurológicos e IRM cerebrales seriadas. Para obtener más información, consultar la sección Histiocitosis de células de Langerhans con síndrome clínico neurodegenerativo.
En niños con HCL en el pulmón, las pruebas de funcionamiento pulmonar y las TC del tórax son métodos sensibles para detectar la progresión de la enfermedad.
En un estudio de 16 años de seguimiento en pacientes de una sola institución, se indicó que los niños con HCL tienen un riesgo más alto de presentar HCL con pulmón de fumador durante la edad adulta, en comparación con otros fumadores jóvenes sanos. La educación continua sobre este riesgo debe formar parte del seguimiento rutinario de los niños con HCL en cualquier sitio.
En resumen, muchos pacientes con enfermedad multisistémica tendrán secuelas a largo plazo debido a la enfermedad subyacente o al tratamiento. Las secuelas endocrinas y del SNC son las más comunes. Estas secuelas a largo plazo afectan de manera significativa la calidad de vida relacionada con la salud en muchos de estos pacientes.[Nivel de evidencia C1] El Children's Oncology Group publicó directrices específicas de seguimiento a largo plazo después del tratamiento del cáncer infantil u otras afecciones con quimioterapia, que están disponibles en su sitio web. Para obtener más información, consultar la sección Enfermedad tardía y efectos del tratamiento de la histiocitosis de células de Langerhans infantil.
Consideraciones especiales para el tratamiento de niños con cáncer
El cáncer en niños y adolescentes es raro, aunque desde 1975 se ha observado un aumento gradual de la incidencia general. Los niños y adolescentes con cáncer se deben derivar a centros médicos que cuenten con equipos multidisciplinarios de especialistas en oncología con experiencia en el tratamiento de los cánceres que se presentan en la niñez y la adolescencia. Este equipo multidisciplinario incorpora la pericia de los siguientes profesionales de atención de la salud y otros para asegurar que los niños reciban el tratamiento, los cuidados médicos de apoyo y la rehabilitación que les permitan lograr una supervivencia y calidad de vida óptimas:
- Médicos de atención primaria.
- Cirujanos pediatra.
- Cirujanos de trasplante.
- Patólogos.
- Radioncólogos pediatras.
- Oncólogos y hematólogos pediatras.
- Oftalmólogos.
- Especialistas en rehabilitación.
- Enfermeros especializados en pediatría.
- Trabajadores o asistentes sociales.
- Profesionales de la vida infantil.
- Psicólogos.
- Nutricionistas y dietistas.
Para obtener información específica sobre los cuidados médicos de apoyo para niños y adolescentes con cáncer, consultar los resúmenes de Cuidados médicos de apoyo y cuidados paliativos.
La American Academy of Pediatrics estableció pautas para los centros de oncología pediátrica y su función en el tratamiento de los pacientes con cáncer infantil. En estos centros de oncología pediátrica, se dispone de ensayos clínicos para la mayoría de los tipos de cáncer que se presentan en niños y adolescentes, y se ofrece la oportunidad de participar a la mayoría de los pacientes y familiares. Por lo general, los ensayos clínicos para los niños y adolescentes con cáncer se diseñan a fin de comparar un tratamiento que parece mejor con el tratamiento estándar actual. La mayoría de los avances en la identificación de tratamientos curativos para los cánceres infantiles se lograron mediante ensayos clínicos. Para obtener información sobre los ensayos clínicos en curso, consultar el portal de Internet del NCI.
Enfermedad de riesgo bajo: Histiocitosis de células de Langerhans monosistémica o multisistémica
Cuadro clínico inicial de la histiocitosis de células de Langerhans monosistémica o multisistémica de riesgo bajo
La presentación clínica más frecuente de la histiocitosis de células de Langerhans (HCL) es una lesión ósea dolorosa; la piel es el segundo órgano afectado en orden de frecuencia. Los síntomas sistémicos de fiebre, pérdida de peso, diarrea, edema, disnea, polidipsia y poliuria se relacionan con un compromiso orgánico específico y un cuadro clínico inicial de enfermedad monosistémica o multisistémica (consultar el Cuadro 2).
| Grupo clínico | Descripción | ||||
|---|---|---|---|---|---|
| SNC = sistema nervioso central; HCL = histiocitosis de células de Langerhans. | |||||
| aReproducción autorizada de Blood, volumen 135, número 16, Carlos Rodríguez-Galindo, Carl E. Allen, Langerhans cell histiocytosis, páginas 1319–1331, derechos de autor 2020, autorizada por Elsevier. | |||||
| Enfermedad multisistémica | Compromiso de 2 o más sistemas | ||||
| Compromiso de órganos de riesgo | Compromiso del hígado, el bazo o la médula ósea | ||||
| Ausencia de compromiso de órganos de riesgo | Ausencia de compromiso del hígado, el bazo o la médula ósea | ||||
| Enfermedad monosistémica | Compromiso de solo 1 sistema | ||||
| Sitio único | Piel, hueso, ganglio linfático u otro sitio (por ejemplo, tiroides o timo) | ||||
| Sitios múltiples | Enfermedad ósea multifocal | ||||
| Sitio especial | Lesión en la base del cráneo con extensión intracraneal o lesión vertebral con extensión al tejido blando intrarraquídeo | ||||
| Histiocitosis de células de Langerhans pulmonar | Enfermedad pulmonar aislada | ||||
| Histiocitosis de células de Langerhans del sistema nervioso central | Lesiones tumorales | ||||
| Enfermedad neurodegenerativa | |||||
| Alteraciones en las imágenes del sistema nervioso central vinculadas con la histiocitosis de células de Langerhans | |||||
| Síntomas en el sistema nervioso central vinculados con la histiocitosis de células de Langerhans | |||||
El compromiso de determinados órganos en el momento del cuadro clínico inicial se usa para clasificar este compromiso como de riesgo alto o de riesgo bajo. El riesgo se refiere al peligro de muerte en los pacientes de riesgo alto. Aunque el compromiso crónico recidivante de órganos de riesgo bajo no suele ser mortal, a veces acarrea consecuencias devastadoras a largo plazo.
- Los órganos de riesgo alto son el hígado, el bazo y el sistema hematopoyético (el compromiso se define por la presencia de al menos 2 alteraciones del linaje en la sangre o por células patológicas positivas para CD1a o CD207 en la médula ósea). Hay nuevos métodos (detección de BRAF V600E mediante PCR o inmunotinción) que permiten una detección más fiable de las células de la HCL en la médula ósea. Los pacientes de riesgo alto suelen ser menores de 2 años. Los pacientes de riesgo alto con compromiso intestinal tienen un riesgo más alto de fracaso terapéutico (un 49 % no responde al tratamiento) que los pacientes sin compromiso intestinal (un 28 % no responde al tratamiento). Sin embargo, la enfermedad intestinal no es un criterio oficial para la enfermedad de riesgo alto.
- Los órganos de riesgo bajo son la piel, los huesos, los pulmones, los ganglios linfáticos, el tubo digestivo, la hipófisis, la tiroides, el timo y el SNC. Se ha descrito compromiso de todos los órganos del cuerpo, salvo los riñones y las gónadas.
Los pacientes con HCL monosistémica (compromiso de un solo órgano) pueden presentar compromiso unifocal (un solo sitio) o multifocal (múltiples sitios). El hueso es el sitio de compromiso de un solo órgano más frecuente. Con menor frecuencia se presenta una HCL multisistémica (compromiso de múltiples órganos) que afecta un número limitado de órganos, o hay enfermedad diseminada. Algunos pacientes con HCL en la piel, los huesos, los ganglios linfáticos o la hipófisis, en cualquier combinación, se clasifican como pacientes de riesgo bajo de muerte, a pesar de un riesgo bastante alto de consecuencias a largo plazo.
Las decisiones de tratamiento dependen del compromiso de órganos de riesgo alto o riesgo bajo y el tipo de presentación de la enfermedad: unifocal, multifocal o multisistémica.
Cuadro clínico inicial de la enfermedad monosistémica de riesgo bajo
La histiocitosis de células de Langerhans (HCL) monosistémica de riesgo bajo afecta solo uno de los siguientes sitios u órganos:
- Huesos.
- Piel y uñas.
- Cavidad oral.
- Ganglios linfáticos y timo.
- Hipófisis.
- Glándula tiroidea.
Huesos
El compromiso óseo es el más común, cerca del 80 % de los pacientes con HCL tienen compromiso en los huesos. La HCL puede afectar cualquier hueso del cuerpo, aunque no suele afectar las manos ni los pies.
Los sitios de las lesiones óseas durante la niñez son los siguientes:
- Lesión lítica del cráneo. El sitio más frecuente de compromiso de la HCL en los niños es una lesión lítica en la bóveda craneal, que puede ser asintomática o dolorosa. A menudo, esta lesión está rodeada por una masa de tejido blando que suele extenderse hacía adentro y afecta la duramadre. Sin embargo, la presencia de esta masa no cambia el pronóstico.
- Fémur, costillas, húmero, pelvis y vértebras. Otros sitios de compromiso esquelético frecuente son el fémur, las costillas, el húmero, la pelvis y las vértebras. Las lesiones en la columna vertebral pueden afectar cualquier vértebra, aunque lo más común es el compromiso de las vértebras cervicales; también es frecuente que las lesiones en la columna se acompañen de otras lesiones óseas. Las lesiones en la columna vertebral en ocasiones producen aplastamiento del cuerpo vertebral (vértebra plana). Las lesiones vertebrales con diseminación al tejido blando suelen ser dolorosas y a veces producen deficiencias neurológicas importantes, Este hallazgo es una indicación para evaluar con una IRM si hay compresión de la médula espinal.
- Huesos de riesgo para el sistema nervioso central. Las lesiones en los huesos faciales, las fosas craneales anterior o media (por ejemplo, temporal, huesos de la órbita, esfenoides, etmoides o cigomas) con extensión tumoral intracraneal comprenden el grupo de riesgo para el SNC. El riesgo de otras enfermedades en el SNC y diabetes insípida se triplica en estos pacientes. Se recomienda tratamiento sistémico para estos pacientes debido al aumento del riesgo de diabetes insípida. La proptosis a causa de una masa de HCL en la órbita se parece a la presentación del rabdomiosarcoma, el neuroblastoma y a los tumores benignos del tejido adiposo del ojo.
Piel y uñas
- Lactantes. El compromiso seborreico del cuero cabelludo a veces se confunde con una costra láctea prolongada en lactantes, a menos que se observe el aspecto purpúrico clásico. El segundo sitio de compromiso más común en esta etapa son los pliegues corporales, como la fosa antecubital y el perineo. Los lactantes también pueden presentar un exantema generalizado, que se parece a muchos otros trastornos cutáneos y no siempre es pruriginoso. Las lesiones cutáneas vesiculares de la HCL se deben diferenciar de las infecciones congénitas.
La HCL cutánea en los lactantes puede circunscribirse a la piel (compromiso cutáneo exclusivo) o acompañarse de compromiso multisistémico. En un informe de 61 casos neonatales entre 1069 pacientes registrados en la base de datos de la Histiocyte Society, casi un 60 % (36 de 61 pacientes) tenía enfermedad multisistémica y, de ellos, un 72 % presentaban compromiso de órganos de riesgo. En un análisis retrospectivo de 71 lactantes y niños con HCL, que tenían compromiso en apariencia exclusivamente cutáneo, se observó que el compromiso multisistémico era más probable en los mayores de 18 meses y que estos pacientes solían recaer después del tratamiento con vinblastina y prednisona. En esta categoría, 8 de 11 pacientes tenían células circulantes con la variante BRAF V600E, en comparación con 1 de 13 pacientes del grupo con compromiso exclusivamente cutáneo. Los pacientes menores de 1 año con compromiso cutáneo exclusivo sometidos a evaluación integral para descartar compromiso de cualquier otro sitio tuvieron una tasa de supervivencia sin progresión a 3 años del 89 % después del tratamiento inicial.
La HCL con compromiso cutáneo exclusivo a veces se resuelve por sí sola y las lesiones desaparecen sin tratamiento durante el primer año de vida. Solo se administra tratamiento cuando los exantemas son muy extensos y hay dolor, ulceración o hemorragia. Estos pacientes se deben controlar de cerca, porque la HCL cutánea exclusiva en neonatos y lactantes muy pequeños puede progresar en cuestión de semanas o meses y convertirse en una enfermedad multisistémica de riesgo alto que pone en peligro la vida.
En una revisión de pacientes que presentaron una HCL con compromiso cutáneo exclusivo en los primeros 3 meses de vida, se compararon los hallazgos clínicos e histopatológicos de 21 niños cuya HCL remitió de manera espontánea con los de 10 niños cuya enfermedad no remitió. Los pacientes cuya enfermedad remitió presentaron lesiones distales que aparecieron durante los primeros 3 meses de vida: pápulas necróticas o máculas hipopigmentadas. Los pacientes con enfermedad que no remitió y que necesitaron tratamiento sistémico presentaron con mayor frecuencia lesiones en áreas intertriginosas. Los estudios inmunohistoquímicos no revelaron diferencias en la expresión de interleucina (IL)-10, Ki-67 o E-cadherina, ni en el número de T-reg, entre los 2 grupos clínicos.
La enfermedad de Hashimoto-Pritzker, o histiocitosis congénita autolimitada con compromiso cutáneo, es una enfermedad de regresión espontánea que muestra el mismo resultado de tinción inmunohistoquímica que la HCL, pero al microscopio electrónico exhibe cuerpos densos que se cree que son mitocondrias senescentes. Una revisión minuciosa de casos originales reveló que algunos pacientes progresaron a HCL multisistémica; se considera que la distinción entre el compromiso cutáneo exclusivo y la enfermedad de Hashimoto-Pritzker carece de valor clínico porque todos estos lactantes se deben observar de cerca después del diagnóstico. Aún no está claro si la presencia o ausencia de la variante BRAF V600E se puede usar para definir la indicación de terapia sistémica para la HCL con compromiso cutáneo exclusivo.
- Niños y adultos. Los niños y los adultos pueden presentar un exantema papular eritematoso, parecido a un exantema candidiásico difuso, en la ingle, el abdomen, la espalda o el tórax. El compromiso seborreico del cuero cabelludo a veces se confunde con un caso grave de caspa en personas mayores. Las lesiones ulcerosas en el cuero cabelludo, detrás de las orejas; en el pliegue mamario inferior; en los genitales; o en la región perianal suelen diagnosticarse de manera errónea como infecciones bacterianas o fúngicas. A veces se observan lesiones vesiculares que deben diferenciarse de lesiones herpéticas.
El compromiso ungueal de la HCL es un hallazgo inusual que se puede presentar en más de un sitio. Suele observarse como estrías longitudinales de color diferente al de las uñas y pérdida de tejido ungueal. Esta afección a menudo responde al tratamiento habitual de la HCL.
Cavidad oral
En la boca, los síntomas iniciales incluyen hipertrofia gingival y úlceras en el paladar blando o duro, la mucosa yugal, o la lengua y los labios. Los dientes hipermóviles (dientes flotantes) y la caída de dientes por lo general indican compromiso del hueso subyacente. Las lesiones en la cavidad oral suelen preceder al compromiso de la HCL en otros sitios.
Ganglios linfáticos y timo
Los ganglios cervicales son los afectados con mayor frecuencia, en grupos apelmazados blandos o duros y acompañados de linfedema. Un timo agrandado, o un compromiso ganglionar mediastínico, a veces se asemeja a un proceso infeccioso y causa síntomas asmáticos. En consecuencia, se indica la biopsia con cultivo para estos casos. El compromiso mediastínico es infrecuente (<5 %) y, a menudo, se presenta con dificultad respiratoria, síndrome de la vena cava superior, o tos y taquipnea. La tasa de supervivencia a 5 años para estos pacientes es del 87 %; las muertes se atribuyen en su mayoría a compromiso hematológico.
Pulmón
En la HCL, el compromiso pulmonar es menos frecuente en los niños que en los adultos porque el hábito de fumar en los adultos es un factor etiológico clave. De 1482 niños del registro francés de HCL, un 7,4 % de los pacientes presentó compromiso pulmonar y un 1 % presentó enfermedad grave que necesitó admisión a cuidados intensivos con inserciones múltiples de tubos de drenaje torácico para varios neumotórax y, en algunos casos, pleurodesis. En una revisión de 178 casos de HCL de otro centro, se encontró compromiso pulmonar en 13 niños (7,3 %), 3 de ellos con enfermedad multisistémica de riesgo alto. El análisis multivariante de la enfermedad pulmonar en la HCL multisistémica no mostró que la enfermedad pulmonar fuera un factor de pronóstico independiente. Las tasas de SG a 5 años fueron del 94 % en aquellos pacientes con compromiso pulmonar y del 96 % en aquellos sin compromiso pulmonar. Rara vez se observa compromiso pulmonar aislado en los niños.
El patrón quístico o nodular de la enfermedad refleja la destrucción del tejido pulmonar inducida por citocinas. En su forma clásica, la enfermedad es simétrica y predomina en los campos pulmonares superiores y medios, no afecta el ángulo costofrénico y produce una imagen muy característica en la TC de alta resolución. La confluencia de quistes a veces lleva a la formación de bullas, y el neumotórax espontáneo en ocasiones es el primer signo de HCL pulmonar, aunque los pacientes pueden presentar taquipnea o disnea. A la larga, la fibrosis generalizada y la destrucción del tejido de los pulmones provocan una insuficiencia pulmonar grave. La disminución de la capacidad de difusión también puede indicar el inicio de hipertensión pulmonar.
La fibrosis generalizada y la reducción de la capacidad de difusión son mucho menos comunes en los niños. En los niños de corta edad con enfermedad difusa, es posible que el tratamiento detenga la progresión de la destrucción tisular y que los mecanismos normales de reparación restauren la función pulmonar, a pesar de que en los estudios radiológicos se sigan observando cicatrices o incluso quistes residuales inactivos.
Hipófisis
Los pacientes con HCL en ocasiones exhiben un compromiso de la parte posterior de la hipófisis y del infundíbulo hipofisario, lo que lleva a diabetes insípida de origen central. El compromiso de la hipófisis anterior a menudo produce retraso del crecimiento y pubertad tardía o precoz. En escasas ocasiones, el compromiso hipotalámico causa obesidad mórbida. Para obtener más información sobre la diabetes insípida, consultar la sección Sistema endocrino.
Glándula tiroidea
Se ha notificado compromiso de la tiroides en la HCL. Los síntomas incluyen agrandamiento tiroideo masivo, hipotiroidismo y síntomas respiratorios.
Cuadro clínico inicial de la enfermedad multisistémica de riesgo bajo
Huesos y otros sistemas orgánicos
Los pacientes con HCL pueden presentar múltiples lesiones óseas como compromiso de un solo órgano (enfermedad ósea monosistémica multifocal) o lesiones óseas acompañadas de compromiso de otros sistemas orgánicos (enfermedad multisistémica con afectación ósea). En un estudio japonés de HCL (JLSG-02) se incluyeron pacientes con compromiso óseo multifocal monosistémico y otros con compromiso multisistémico que incluyó el hueso. En una revisión del estudio se encontró que los pacientes del grupo de enfermedad multisistémica con compromiso óseo tenían más probabilidades de presentar lesiones en el hueso temporal, la apófisis mastoides, el peñasco, la órbita y el hueso cigomático (es decir, huesos de riesgo para el SNC). Estos pacientes también tuvieron una incidencia más alta de diabetes insípida, que se correlacionó con una mayor frecuencia de lesiones óseas de riesgo. En un estudio de la Histiocyte Society se encontró una disminución de la mortalidad en pacientes con HCL multisistémica de riesgo alto con compromiso óseo, lo que indica que la HCL ósea quizás tenga una evolución más lenta.
Órganos abdominales y aparato digestivo
En la HCL, el hígado y el bazo son órganos de riesgo alto, y el compromiso de estos órganos afecta el pronóstico. Para obtener más información, consultar las secciones Hígado (colangitis esclerosante) y Bazo.
Aunque es poco frecuente, se notificó infiltración de la HCL en el páncreas y los riñones.
Los pacientes presentan diarrea, rectorragia, fístulas perianales o síndrome de malabsorción.
Sistema endocrino
La diabetes insípida, causada por el daño de la HCL a las células que secretan la hormona antidiurética en la hipófisis posterior, es la manifestación endocrina más frecuente de la HCL. Por lo general, en la IRM se observa nodularidad o engrosamiento del tallo hipofisario y pérdida del punto brillante de la hipófisis en las imágenes ponderadas en T2. Cuando hay engrosamiento del infundíbulo hipofisario, o este es muy grande, hay una probabilidad del 50 % de que el paciente tenga un germinoma, una HCL o un linfoma. Las biopsias de la hipófisis son muy infrecuentes, pero a veces se indican cuando es el único sitio de enfermedad y el grosor del infundíbulo hipofisario es de más de 6,5 mm o hay una masa hipotalámica. Si la enfermedad hipofisaria se asocia con otros sitios de compromiso, es posible obtener una biopsia de esos sitios para establecer el diagnóstico.
Alrededor del 4 % de los pacientes con HCL presentan una diabetes insípida en apariencia idiopática antes de que se identifique la presencia de otras lesiones de la HCL. En un estudio prospectivo de seguimiento se inscribieron pacientes pediátricos con diabetes insípida central idiopática como cuadro clínico inicial que solo recibieron tratamiento para la diabetes insípida. En el estudio se observó que el 19 % de los pacientes, con el tiempo, presentaron signos de HCL, mientras que en el 18 % se diagnosticó craneofaringioma y en el 10 % germinoma. En un estudio prospectivo de las causas de la diabetes insípida de origen central en niños y adultos jóvenes, se encontró que el 15 % de los pacientes tenían HCL, el 11 % presentaban germinomas y el 7 % tenían craneofaringiomas. Los otros diagnósticos se relacionaron con traumatismos, asociación familiar o defectos de la línea media, y el 50 % permanecieron con estado idiopático. Las decisiones sobre el tratamiento de un paciente con diabetes insípida de origen central aparentemente aislada, o de un paciente con HCL sin confirmación por biopsia, siguen siendo controvertidas.
El abordaje es diferente para los pacientes con HCL confirmada que se acompaña de diabetes insípida. Estos pacientes tienen entre un 50 % y un 80 % más de probabilidades de presentar otras lesiones diagnósticas de HCL (incluso lesiones óseas, pulmonares o cutáneas) durante el primer año luego de la aparición de la diabetes insípida. En general, es más común que los pacientes con HCL presenten diabetes insípida en un momento más tardío durante la evolución de la enfermedad, según se destaca en los siguientes estudios:
- En un estudio se comparó la incidencia de diabetes insípida en pacientes que no habían recibido tratamiento sistémico y en pacientes que recibieron 6 meses de vinblastina y prednisona. Los pacientes que no recibieron tratamiento sistémico tuvieron una incidencia de diabetes insípida del 40 %. Los pacientes que recibieron quimioterapia presentaron una incidencia de diabetes insípida del 20 %. Este hallazgo apoya en gran medida el tratamiento de las lesiones óseas de riesgo para el SNC, incluso cuando la enfermedad se restringe a un solo sitio óseo.
- En un estudio de 589 pacientes con HCL, el riesgo a 10 años de compromiso hipofisario fue del 24 %. La diabetes insípida se observó al cabo de una media de 1 año después del diagnóstico de HCL. Entre los pacientes con HCL y diabetes insípida, el 56 % presentó deficiencias de la hipófisis anterior (hormonas del crecimiento, tirodoestimulante y gonadoestimulante) en el transcurso de 10 años desde el inicio de la diabetes insípida. En este estudio, no se observó disminución en la incidencia de diabetes insípida en los pacientes tratados con quimioterapia, pero esto puede reflejar la duración del tratamiento o el número de fármacos utilizados.
- El grupo alemán-austriaco-neerlandés (Deutsche Arbeitsgemeinschaft für Leukämieforschung und Behandlung im Kindesalter [DAL]) administró terapia durante más tiempo y con más fármacos quimioterapéuticos y encontró una incidencia acumulada de diabetes insípida del 20 % a los 15 años del diagnóstico de HCL. La incidencia de diabetes insípida también fue más baja en los pacientes tratados con regímenes quimioterapéuticos más intensivos en los estudios HISTSOC-LCH-III (NCT00276757), JLSG-96 y JLSG-02 de Japón (8,9 % para enfermedad multisistémica), en comparación con los estudios HISTSOC-LCH-I e HISTSOC-LCH-II (14,2 %). En general, la diabetes insípida se presentó en el 11 % de los pacientes tratados con quimioterapia multifarmacológica y en hasta el 50 % de los pacientes tratados de forma menos radical.
En el momento del diagnóstico, los pacientes con enfermedad multisistémica y compromiso craneofacial (en particular, órbita, mastoides y temporal) presentaron un aumento significativo en el riesgo de desarrollar diabetes insípida durante el curso de la enfermedad (riesgo relativo, 4,6). De los pacientes con HCL y diabetes insípida, el 75 % presentaban estas lesiones óseas de riesgo para el SNC. El riesgo de diabetes insípida aumenta cuando la HCL se mantiene activa durante un período más largo o se reactiva.
Cerca del 50 % de los pacientes que presentan diabetes insípida aislada (como manifestación inicial de la HCL) tienen alteraciones en la hipófisis anterior en el momento del diagnóstico o las presentan durante los 10 años posteriores al inicio de la diabetes insípida. Las alteraciones de la hipófisis anterior incluyen amenorrea secundaria, panhipopituitarismo, deficiencia de la hormona del crecimiento, hipoadrenalismo y alteraciones de las gonadotropinas. La incidencia de alteraciones de la hipófisis anterior es más alta en los pacientes con HCL que en aquellos con diabetes insípida central idiopática verdadera.
Ocular
Aunque es infrecuente, se ha notificado HCL ocular, que a veces produce ceguera. Es posible que otros sistemas orgánicos estén comprometidos y que la HCL ocular no responda bien a la quimioterapia convencional.
Sistema nervioso central
Manifestaciones de la enfermedad en el sistema nervioso central
Los pacientes con HCL a veces presentan lesiones expansivas en la región hipotálamo-hipofisaria, el plexo coroideo, la sustancia gris o la sustancia blanca. Estas lesiones contienen células de la HCL positivas para CD1a y linfocitos positivos para CD8 y, por lo tanto, son lesiones activas de la HCL.
Los pacientes con tumores hipofisarios grandes (>6,5 mm) presentan un riesgo más alto de disfunción de la hipófisis anterior y HCL neurodegenerativa en el sistema nervioso central (SNC). En un estudio retrospectivo de 22 pacientes, se encontró que todos presentaban signos radiológicos de HCL neurodegenerativa en el SNC detectados al cabo de una mediana de tiempo de 3 años y 4 meses después del diagnóstico de HCL; dichos signos radiológicos empeoraron en 19 pacientes. Se observó disfunción neurológica en 5 pacientes, disfunción de la hipófisis anterior en 18 de 22 pacientes y diabetes insípida en 20 pacientes. Se presentó deficiencia de la hormona del crecimiento en 21 pacientes, deficiencia de la hormona luteinizante o la hormona foliculoestimulante en 10 pacientes y deficiencia de la hormona tiroidea en 10 pacientes.
Histiocitosis de células de Langerhans con síndrome clínico neurodegenerativo
Del 1 % al 4 % de los pacientes con histiocitosis de células de Langerhans presentan un síndrome clínico neurodegenerativo crónico llamado HCL-cND. Estos pacientes a veces tienen temblores, alteraciones en la marcha, ataxia, disartria, cefaleas, alteraciones visuales, problemas cognitivos y conductuales, y psicosis.
De 1897 pacientes con HCL, 36 pacientes recibieron un diagnóstico de histiocitosis de células de Langerhans con HCL-cND. La incidencia de HCL-cND fue del 4,1 % a los 10 años de seguimiento. Este subtipo fue más frecuente en los pacientes con compromiso hipofisario (86,1 vs. 12,2 % sin lesiones hipofisarias), compromiso cutáneo (75 vs. 34,2 % sin lesiones cutáneas), y compromiso óseo de la base del cráneo (63,9 vs. 28,4 % sin lesiones del cráneo). Fue más probable que los pacientes con una variante de BRAF presentaran una HCL-cND (93,7 %) que los que no tenían la variante (54,1 %). En el análisis multivariante, el riesgo general de HCL-cND fue de 2,13 en los pacientes con lesiones en la base del cráneo, de 9,8 en pacientes con la variante BRAF V600E, y de 30,88 para los pacientes con compromiso hipofisario. El riesgo de HCL-cND no se había estabilizado hasta 20 años después del diagnóstico de HCL.
En las IRM encefálicas de estos pacientes se observa una señal hiperintensa en el núcleo dentado y la sustancia blanca del cerebelo en las imágenes ponderadas en T2, o se observan lesiones hiperintensas en los núcleos basales en las imágenes ponderadas en T1 o atrofia del cerebelo. Los hallazgos radiológicos pueden preceder a la aparición de los síntomas por muchos años o se encuentran de manera coincidente. Se publicó un estudio de 83 pacientes con HCL que se sometieron a por lo menos 2 IRM encefálicas para evaluar lesiones craneofaciales, diabetes insípida u otras deficiencias endocrinas relacionadas con síntomas neuropsicológicos. De 83 pacientes, 47 (57 %) presentaron cambios neurodegenerativos radiológicos tras una mediana de tiempo de 34 meses desde el diagnóstico de HCL. Se presentaron deficiencias neurológicas clínicas en 12 (25 %) de los 47 pacientes entre 3 y 15 años después del diagnóstico de HCL y deficiencias casi imperceptibles en la memoria auditiva a corto plazo en 14 de 47 pacientes.
En la evaluación histológica inicial de estas lesiones neurodegenerativas, se notificó una infiltración abundante de células T, por lo general sin células dendríticas positivas para CD1a, además de activación microglial y astrocitosis. Sin embargo, en un informe de 2018, en el análisis de tejido encefálico de pacientes con HCL con síndrome clínico neurodegenerativo se encontró infiltración perivascular de células negativas para CD207 pero que reaccionaron a la tinción para la proteína alterada BRAF V600E en la protuberancia, el cerebelo y los núcleos basales. Estas son áreas que se identifican mediante hallazgos anormales en las IRM que son característicos de las imágenes con recuperación de la inversión atenuada de fluido (FLAIR) en T2. El análisis con PCR cuantitativa de estas áreas indicó aumento en el número de células con alteraciones de BRAF y una expresión elevada de osteopontina. El tejido encefálico en estas áreas mostró desmielinización activa, que se correlacionó con los hallazgos radiológicos y el deterioro clínico.
Se publicó un estudio sobre las consecuencias permanentes en el SNC (deficiencias neuropsicológicas) en 14 de 25 pacientes con HCL observados durante una mediana de 10 años. De estos pacientes, 7 tenían diabetes insípida y 5 tenían evidencia radiográfica de cambios neurodegenerativos de HCL en el SNC. Los pacientes con lesiones craneofaciales tuvieron puntuaciones más bajas de rendimiento y CI verbal que los pacientes con otras lesiones de HCL.
Tratamiento de la histiocitosis de células de Langerhans monosistémica o multisistémica de riesgo bajo
Durante varios años, los grupos de estudio nacionales e internacionales han definido los grupos de tratamiento según el riesgo de manera que los pacientes con histiocitosis de células de Langerhans (HCL) se asignan en función del riesgo de muerte y de efectos tardíos por la enfermedad.
Según el sitio y la extensión de la enfermedad, es posible que el tratamiento de la HCL incluya observación (después de la biopsia o el curetaje), cirugía, radioterapia o fármacos por vía oral, tópica o intravenosa. La duración recomendada para el tratamiento es de 12 meses en los pacientes que necesitan quimioterapia cuando la enfermedad es monosistémica con compromiso óseo, cutáneo o ganglionar.
En los pacientes con enfermedad multisistémica de riesgo alto o bajo, la tasa de reactivación después de 6 meses de tratamiento fue de hasta el 50 % en los ensayos HISTSOC-LCH-I e HISTSOC-LCH-II. En los ensayos del grupo alemán-austriaco-neerlandés (Deutsche Arbeitsgemeinschaft für Leukämieforschung und Behandlung im Kindesalter [DAL]) se trataron a los pacientes durante 1 año y se presentaron menos recaídas (29 %). A partir de estos hallazgos, el ensayo HISTSOC-LCH-III se diseñó para administrar 12 meses de quimioterapia a todos los pacientes con enfermedad multisistémica de riesgo alto y para asignar al azar a recibir 6 o 12 meses de tratamiento a los pacientes con enfermedad multisistémica de riesgo bajo. La tasa de reactivación se redujo significativamente hasta un 30 % en los pacientes con enfermedad de riesgo bajo o riesgo alto que recibieron 12 meses de tratamiento.
El tratamiento estándar para la HCL se basa en datos de ensayos internacionales con un gran número de pacientes. Sin embargo, algunos pacientes tienen HCL solo en la piel, la boca, la hipófisis u otros sitios que no se estudiaron en estos ensayos internacionales. En esas situaciones, las recomendaciones terapéuticas se fundamentan en series de casos que no cuentan con la solidez de la evidencia obtenida de los ensayos.
La Histiocyte Society organiza ensayos clínicos de tratamiento pediátrico en los que se han inscrito pacientes desde la década de 1980. La información sobre los centros que están inscribiendo pacientes en estos ensayos se encuentra en el portal de Internet ClinicalTrials.gov.
Las opciones de tratamiento para los pacientes con enfermedad monosistémica o multisistémica de riesgo bajo varían según el sitio de compromiso, como se explica a continuación:
- Compromiso cutáneo aislado.
- Compromiso esquelético.
- Enfermedad en el sistema nervioso central.
Compromiso cutáneo aislado
Las opciones de tratamiento para los pacientes con compromiso cutáneo aislado son las siguientes:
- Observación. Se recomienda la observación para todos los pacientes pediátricos con HCL asintomática solo en la piel.
- Tratamiento. Se recomienda administrar tratamiento solo en pacientes con enfermedad sintomática, como los que presentan exantemas extensos, dolores, ulceraciones o hemorragias.
Los pacientes con compromiso cutáneo exclusivo necesitan una evaluación completa para la estadificación debido a que se encontró enfermedad multisistémica que exigía tratamiento en un 41 % de estos pacientes remitidos a un centro de atención. Se indica un seguimiento clínico cuidadoso (pero no radiológico) de los lactantes pequeños con HCL solo en la piel porque es posible que la enfermedad progrese a un compromiso multisistémico de riesgo alto. Los niños pequeños con HCL solo en la piel se deben vigilar periódicamente durante muchos años porque 1 de 19 niños y 1 de 25 niños en dos series presentaron diabetes insípida tardía.
Las opciones de tratamiento para los pacientes con lesiones cutáneas aisladas y sintomáticas son las siguientes:
- Corticoesteroides tópicos. Los corticoesteroides de potencia media a alta son eficaces, pero la recidiva después de la interrupción es común.
- Metotrexato oral. Metotrexato oral (20 mg/m2) semanal durante 6 a 12 meses.
- Hidroxiurea oral. Hidroxiurea oral (20 mg/kg) diaria durante por lo menos 12 meses.
- Talidomida o lenalidomida orales. Talidomida oral de 50 mg a 200 mg cada noche. La talidomida o lenalidomida orales pueden ser eficaces tanto para pacientes pediátricos como adultos.
- Mostaza nitrogenada de uso tópico. La aplicación tópica de mostaza nitrogenada puede ser eficaz para la HCL cutánea resistente a las terapias orales, pero no para la enfermedad que afecta grandes áreas de la piel.
- Psoraleno y radiación ultravioleta A de onda larga (PUVA) o UVB. El psoraleno y la PUVA o la UVB pueden ser eficaces para la HCL cutánea, pero su uso está limitado por la posibilidad de cánceres de piel tardíos, en especial en pacientes de piel clara.
- Quimioterapia. Es posible tratar los casos graves y sintomáticos con quimioterapia sistémica.
- Radioterapia. Aunque se ha utilizado radioterapia de haz externo, no se ha demostrado eficacia uniforme y puede tener efectos secundarios graves.
Compromiso esquelético
Lesiones craneales únicas en las regiones frontal, parietal u occipital, o lesiones únicas en cualquier otro hueso
Las opciones de tratamiento para los pacientes con lesiones craneales únicas en las regiones frontal, parietal u occipital, o con lesiones únicas en cualquier otro hueso, son las siguientes:
- Curetaje. El curetaje solo es el tratamiento recomendado, cuando es posible, para las lesiones óseas aisladas. También se puede usar curetaje con inyección de metilprednisolona. Tal vez no se indique la extirpación completa de las lesiones óseas de la HCL porque esto aumenta el tiempo de cicatrización y el riesgo de complicaciones a largo plazo. No es necesaria la extirpación completa de las lesiones craneales, que a veces exige el uso de injertos.
- Radioterapia de dosis baja. Se podría considerar la radioterapia local para una lesión aislada.[Nivel de evidencia C3] La radioterapia de dosis bajas (7–10 Gy) es eficaz, pero su uso se limita en pacientes pediátricos solo para las lesiones que amenazan el funcionamiento de los órganos o que son dolorosas y no susceptibles de otras terapias.; [Nivel de evidencia C1] En un estudio de una sola institución con 39 pacientes de HCL (intervalo de edad, 1,5–67 años; 15 pacientes de <18 años) que recibieron radioterapia para 46 lesiones, no hubo recidivas locales en los 31 sitios óseos (mediana de dosis de radioterapia, 10,8 Gy; intervalo, 7,5–24 Gy) y la tasa de ausencia de fracaso local fue del 63 % a los 3 años en las 15 lesiones no óseas (intervalo de confianza 95 %, 32–83 %; P = 0,0008). En este estudio, no se presentaron cánceres subsiguientes; aunque se han notificado cánceres subsiguientes en otros casos. Las complicaciones esqueléticas son poco frecuentes después de las dosis bajas que se usan, pero pueden presentarse.
Lesiones craneales en apófisis mastoides, temporal y órbita
Los huesos de riesgo para el SNC incluyen las apófisis mastoides, los huesos temporales, el esfenoides, los cigomáticos, el etmoides, los maxilares, los huesos de la órbita, los huesos de los senos paranasales y los huesos que forman la fosa craneal anterior o media. Este riesgo se refiere al aumento del riesgo de progresión a diabetes insípida cuando se produce el compromiso encefálico (SNC).
El propósito del tratamiento de los pacientes con lesiones aisladas de riesgo para el SNC es disminuir la probabilidad de presentar diabetes insípida y otros problemas neurológicos a largo plazo.
Las opciones de tratamiento para los pacientes con lesiones craneales en apófisis mastoides, temporal u órbita son las siguientes:
- Quimioterapia. El tratamiento actual para los huesos de riesgo para el SNC es 12 meses de vinblastina y prednisona, según el estudio HISTSOC-LCH-III (NCT00276757):[Nivel de evidencia B1]
- Vinblastina semanal (6 mg/m2) durante 7 semanas para una respuesta adecuada.
- Prednisona diaria (40 mg/m2) durante 4 semanas y después reducción gradual durante 2 semanas.
- A continuación, se administra prednisona durante 5 días en dosis de 40 mg/m2 cada 3 semanas acompañada de inyecciones de vinblastina (también cada 3 semanas).
Hay polémica sobre la necesidad de tratamiento sistémico durante el cuadro clínico inicial de una HCL ósea unifocal, incluso cuando hay compromiso de huesos de riesgo para el SNC. En una revisión retrospectiva se notificó sobre una serie de pacientes con lesiones en órbitas o mastoides que se sometieron solo a curetaje quirúrgico. El tratamiento lo completó un solo cirujano especializado en otorrinolaringología. Ninguno de estos pacientes presentó diabetes insípida.
Sin embargo, al comparar las tasas de incidencia de diabetes insípida en pacientes que recibieron poca quimioterapia, o que no la recibieron (incidencia del 20–50 %), con las tasas de incidencia notificadas en el ensayo DAL-HX 83 del grupo alemán-austriaco-neerlandés (incidencia del 10 % en pacientes tratados por HCL), se observó que la solidez de la evidencia del ensayo DAL-HX 83 respalda el tratamiento quimioterapéutico para prevenir la diabetes insípida en pacientes con HCL en huesos de riesgo para el SNC. No obstante, cabe destacar que en los estudios DAL-HX se usaron más fármacos y el tratamiento duró 12 meses.
Lesiones óseas vertebrales o femorales en riesgo de fractura
Las opciones de tratamiento para pacientes con lesiones óseas vertebrales o femorales en riesgo de fractura son las siguientes:
- Observación. Es posible observar una lesión de cuerpo vertebral cuando no se ha extendido al tejido blando que rodea el espacio extradural.
- Radioterapia de dosis baja. Es posible usar dosis bajas de radioterapia para promover la resolución de una lesión aislada de un cuerpo vertebral o una lesión grande del cuello femoral con riesgo de fractura, para las que no se suele indicar quimioterapia (lesión ósea única). A pesar de la dosis baja requerida (7–10 Gy), la radioterapia se debe usar con precaución debido a las preocupaciones sobre neoplasias malignas secundarias en los tejidos adyacentes, deformidades esqueléticas cuando se irradian las placas de crecimiento de niños muy pequeños o cuando la tiroides se encuentra en el campo de radiación de las lesiones vertebrales cervicales.
- Quimioterapia. Los pacientes con extensión de las lesiones vertebrales al tejido blando a menudo responden bien al tratamiento con quimioterapia,[Nivel de evidencia C2] pero no necesitan prolongar el tratamiento después del período necesario para reducir la masa y controlar el riesgo para la médula espinal. El riesgo de reactivación de una sola lesión ósea fue de solo un 9 % en una serie retrospectiva grande.
- Ortesis o artrodesis raquídea. Cuando hay inestabilidad de las vértebras cervicales o síntomas neurológicos, es posible que se necesite utilizar refuerzo por ortesis y en casos poco frecuentes una artrodesis raquídea.
Lesiones óseas múltiples (lesiones óseas monosistémicas multifocales)
Las opciones de tratamiento para pacientes con lesiones óseas múltiples (lesiones óseas monosistémicas multifocales) en riesgo de fractura son las siguientes:
- Quimioterapia. El régimen de quimioterapia sistémica más común es la combinación de vinblastina y prednisona. A partir de los resultados del ensayo HISTSOC-LCH-III (NCT00276757), se administra un tratamiento de 12 meses con vinblastina semanal (6 mg/m2 ) durante 7 semanas y luego cada 3 semanas para quienes respondan bien. La prednisona (40 mg/m2) se administra todos los días durante 4 semanas y luego se reduce durante 2 semanas. Después, se administra prednisona durante 5 días en dosis de 40 mg/m2 cada 3 semanas, acompañada de inyecciones de vinblastina.
Un ciclo corto (<6 meses) de monoterapia (por ejemplo, prednisona) no es suficiente y el número de recaídas es más alto. Se informó de una tasa de reactivación del 18 % cuando se usó un régimen multifarmacológico durante 6 meses versus una tasa de reactivación histórica del 50 % al 80 % con cirugía sola o con un régimen de monoterapia. En una comparación de los resultados de dos ensayos realizados en Japón, no se observó mejoría en las tasas de supervivencia sin progresión (66 vs. 65 %) cuando se añadió más prednisona y una fase de mantenimiento prolongada.
Para obtener información sobre otros fármacos que se usan para tratar la HCL ósea multifocal, consultar la sección Lesiones óseas múltiples en combinación con compromiso cutáneo, compromiso ganglionar o diabetes insípida (HCL multisistémica de riesgo bajo).
Lesiones óseas múltiples en combinación con compromiso cutáneo, compromiso ganglionar o diabetes insípida (HCL multisistémica de riesgo bajo)
Las opciones de tratamiento para las lesiones óseas múltiples en combinación con compromiso cutáneo, compromiso ganglionar o diabetes insípida (HCL multisistémica de riesgo bajo) son las siguientes:
- Quimioterapia (combinación de vinblastina y prednisona). A partir de los resultados del ensayo aleatorizado HISTSOC-LCH-III (NCT00276757), se administra el mismo régimen quimioterapéutico de vinblastina y prednisona durante 12 meses, como se describió en la sección anterior. Los pacientes sin compromiso de órganos de riesgo que se asignaron al azar a 12 meses de vinblastina y prednisona tuvieron una tasa más baja de reactivación a 5 años (37 %) que los pacientes que recibieron solo 6 meses de tratamiento (54 %; P = 0,03) y los pacientes que se trataron con programas tradicionales de 6 meses (52 % [HISTSOC-LCH-I] y 48 % [HISTSOC-LCH-II]; P< 0,001). La mayoría de las reactivaciones de la enfermedad se produjeron en huesos, piel u otras ubicaciones sin riesgo.
Los pacientes con HCL multisistémica de riesgo bajo tienen una tasa de supervivencia de casi el 100 %, pero las reactivaciones mostraron ser factores de riesgo importantes de efectos tardíos significativos en los ensayos del DAL y de la Histiocyte Society.
- Quimioterapia (otros regímenes). Otros regímenes de quimioterapia que también han resultado eficaces son los siguientes:
- Combinación de vincristina, citarabina y prednisona.[Nivel de evidencia C2] Esta combinación ha demostrado eficacia como terapia de primera línea o de rescate. Sin embargo, ahora se administra prednisona durante un período mucho más corto que el tiempo publicado en un principio (52 semanas): 40 mg/m2 por 4 semanas, luego se reduce a 20 mg/m2 en la semana 6 durante la fase de inducción, y se sigue durante 5 días cada 3 semanas en dosis de 20 mg/m2 combinada con una sola dosis de vincristina y 5 días de citarabina durante el mantenimiento.
- Cladribina. La cladribina en dosis de 5 mg/m2 al día por 5 días cada 3 semanas durante 2 a 6 ciclos puede ser una terapia de rescate eficaz para la enfermedad ósea recidivante o multisistémica de riesgo bajo.[Nivel de evidencia C2] No se recomiendan más de 6 ciclos debido al riesgo de citopenias acumuladas.
- Terapia con bisfosfonatos. La terapia con bisfosfonatos también puede ser eficaz para tratar las lesiones óseas de la HCL.[Nivel de evidencia C2] En una encuesta nacional de Japón, se caracterizó a 16 niños tratados con bisfosfonatos por una HCL ósea. Ninguno tenía enfermedad en órganos de riesgo. La mayoría de los pacientes recibió 6 ciclos de pamidronato en dosis de 1 mg/kg por curso de tratamiento administrados a intervalos de 4 semanas. En 12 de 16 pacientes se resolvieron todas las lesiones activas, incluso las lesiones cutáneas (n = 3) y de tejidos blandos (n = 3). Al cabo de una mediana de 3,3 años, 8 pacientes permanecieron sin enfermedad. Otros bisfosfonatos, como el zoledronato, se han utilizado para tratar con éxito la HCL ósea.
Aunque los bisfosfonatos se usan para la HCL ósea, en algunas publicaciones se notifican respuestas en otros órganos, como la piel.
Enfermedad en el sistema nervioso central
Lesiones en el sistema nervioso central
Las lesiones de la HCL en el SNC son las siguientes:
- Lesiones expansivas o tumores en el cerebro, el cerebelo o el plexo coroideo.
- Lesiones expansivas en el eje hipotalámico-hipofisario que siempre se relacionan con la diabetes insípida y, a menudo, con otras endocrinopatías.
Se usan fármacos que cruzan la barrera hematoencefálica, como la cladribina y otros análogos nucleosídicos, por ejemplo, la citarabina, para las lesiones activas de la HCL del SNC.
Las opciones de tratamiento para los pacientes con lesiones de HCL en el SNC son las siguientes:
- Quimioterapia (cladribina). El tratamiento de las lesiones compresivas con cladribina ha sido eficaz en 13 casos notificados.; [Nivel de evidencia C2] Estas lesiones fueron agrandamiento del eje hipotalámico-hipofisario, lesiones compresivas parenquimatosas y compromiso leptomeníngeo. Las dosis de cladribina oscilaron entre 5 mg/m2 y 13 mg/m2, administradas en diferentes frecuencias.[Nivel de evidencia C2]
- Quimioterapia (otros regímenes). Los pacientes con HCL y lesiones compresivas en la región hipotálamo-hipofisaria, el plexo coroideo, la sustancia gris o la sustancia blanca también pueden responder a la quimioterapia estándar para la HCL.[Nivel de evidencia C3] El tratamiento de vinblastina con corticoesteroides o sin estos en pacientes con lesiones compresivas en el SNC (20 pacientes; principalmente hipofisarias) produjo respuestas objetivas en 15 pacientes. De 20 pacientes, 5 lograron respuestas completas y 10 obtuvieron respuestas parciales.
Histiocitosis de células de Langerhans con síndrome clínico neurodegenerativo
No se ha establecido ningún tratamiento óptimo para la HCL-cND y la evaluación de la respuesta es difícil.
En la HCL-cND se presentan señales hiperintensas en las imágenes FLAIR en T2, con mayor frecuencia, en la sustancia blanca cerebelosa, la protuberancia, los núcleos basales y, algunas veces, en el cerebro. No queda claro si se deben tratar los cambios de la HCL que se observan en las IRM en el cerebelo, la protuberancia y los núcleos basales, cuando estos no se acompañan de manifestaciones clínicas a nivel neurológico. En los estudios iniciales se observó que no todos los cambios radiológicos relacionados con la HCL progresaron a una enfermedad clínica neurodegenerativa. Sin embargo, es importante establecer un tratamiento en los estadios tempranos de la enfermedad clínica antes de que aparezca el daño permanente. La recomendación actual es la vigilancia con evaluación neurológica, tanto clínica como con IRM. El tratamiento comienza tan pronto como se observa la progresión clínica de la enfermedad neurodegenerativa. No está claro si los cambios radiológicos progresivos deben ser una indicación para comenzar el tratamiento.
Se han utilizado otros fármacos para la HCL activa, como la dexametasona, la cladribina y el infliximab, en un número pequeño de pacientes con resultados contradictorios. Muchos de estos fármacos pueden dar lugar a la resolución completa o parcial de los hallazgos radiográficos, pero las tasas de respuesta clínica definitiva no se han definido de manera rigurosa.; [Nivel de evidencia C2]
Las opciones de tratamiento para los pacientes con HCL-cND son las siguientes:
- Terapia con inhibidores de BRAF V600E. Para obtener más información, consultar la sección Tratamiento de la histiocitosis de células de Langerhans multisistémica recidivante, resistente al tratamiento o progresiva de riesgo alto.
La experiencia clínica indica que la terapia con inhibidores de BRAF V600E quizás sea la opción más eficaz para mejorar los síntomas neurológicos en la HCL-CND, pero es posible que este tratamiento deba continuarse de por vida.[Nivel de evidencia C3];
- Quimioterapia. En un estudio en el que se usó citarabina con vincristina o sin esta durante un máximo de 24 meses, se notificaron mejoras en los hallazgos clínicos y de IRM en algunos pacientes y enfermedad estable en otros.[Nivel de evidencia C1] Se vigiló a 7 de 8 pacientes durante más de 8 años después de suspender el tratamiento y se observaron hallazgos neurológicos y radiográficos estables.
En el protocolo JLSG-96 del Japan LCH Study Group, la citarabina no logró prevenir el inicio del síndrome neurodegenerativo. Los pacientes recibieron citarabina en dosis de 100 mg/m2 diariamente los días 1 a 5 durante la inducción y 150 mg/m2 el día 1 de cada ciclo de mantenimiento (cada 2 semanas durante 6 meses). De 91 pacientes, 13 presentaron enfermedad neurodegenerativa, que es similar a la tasa notificada en los pacientes en los estudios de la Histiocyte Society.[Nivel de evidencia B4]
- Rituximab. Se trató con rituximab a 8 pacientes con síntomas neurológicos durante una mediana de 8 años y que presentaron síntomas nuevos después de recibir tratamiento con citarabina (375 mg/m2 semanal durante 4 semanas, repetidos cada 3 meses y aumentados a 555 mg/m2 sin mejoría o empeoramiento de los síntomas) por períodos de tiempo variables. Los síntomas clínicos mejoraron en 7 de 8 pacientes (5 pacientes mejoraron al cabo de 1 mes desde el inicio del rituximab). Cinco pacientes permanecieron sin síntomas clínicos progresivos durante 3 años o más.[Nivel de evidencia C3]
El reconocimiento precoz de la neurodegeneración clínica y el establecimiento de tratamiento temprano es esencial para el éxito terapéutico. En varios países se encuentran en curso estudios en los que se combinan los hallazgos de las IRM y los marcadores de la desmielinización en el líquido cefalorraquídeo (LCR), para identificar a los pacientes que necesitan tratamiento, incluso antes de que aparezcan síntomas clínicos. También se están realizando estudios sobre el uso del LCR y biomarcadores séricos para predecir y prevenir la enfermedad neurodegenerativa.
Histiocitosis de células de Langerhans multisistémica de riesgo alto
Cuadro clínico inicial de la histiocitosis de células de Langerhans multisistémica de riesgo alto
Hígado (colangitis esclerosante)
Es posible que el hígado esté agrandado debido a infiltración directa de las células de la HCL, o como reacción secundaria al exceso de citocinas, que provoca activación de macrófagos o infiltración de linfocitos alrededor de las vías biliares. Las células de la HCL presentan tropismo portal (vías biliares) que a veces lleva a lesión biliar y esclerosis ductal. Es posible que también se encuentren células de la HCL peribiliares y, en raras ocasiones, masas nodulares de la HCL.
Cuando la HCL afecta el hígado, se observan áreas hipoecogénicas o una señal de baja intensidad a lo largo del sistema portal o las vías biliares en las ecografías, las TC o las IRM del hígado. Si bien la ecografía o la colangiografía por IRM pueden ser útiles para el diagnóstico de esta complicación, la biopsia hepática es la única forma definitiva de corroborar una HCL activa o una fibrosis hepática residual. Los resultados de la biopsia a menudo muestran linfocitos y efectos obstructivos biliares sin células de la HCL.
Los pacientes con HCL hepática exhiben hepatomegalia (>3 cm por debajo del margen costal en la línea medioclavicular) o hepatoesplenomegalia y disfunción, comprobadas por hipoproteinemia (<55 g/l, hipoalbuminemia <25 g/l) o hallazgos histológicos de enfermedad activa. Los pacientes también pueden presentar concentraciones elevadas de fosfatasa alcalina, transaminasas hepáticas y γ-glutamil–transpeptidasa, así como disfunción de la coagulación o ascitis.
Las complicaciones más graves de la HCL hepática son la colestasis y la colangitis esclerosante. Por lo general, esto ocurre meses después de la presentación inicial, pero en ocasiones se observa en el momento del diagnóstico. La mediana de edad de los niños con esta forma de HCL hepática es de 23 meses. La historia natural de la colangitis esclerosante es variable. Algunos pacientes que reciben quimioterapia mejoran, mientras que otros pacientes tienen enfermedad estable o progresan desde esclerosis hasta cirrosis biliar e hipertensión portal, que se puede observar incluso en ausencia de células de la HCL activas. En un informe de 13 pacientes con HCL y hepatopatía, se encontró que todos los pacientes presentaban variantes BRAF V600E en las muestras de biopsias de piel, huesos o hígado. No se sabe si el uso temprano de la terapia con inhibidores en este grupo de pacientes reducirá o evitará la progresión de la colangitis esclerosante. Esta terapia aún debe investigarse.
Bazo
Esplenomegalia masiva (por lo general, >2 cm por debajo del margen costal en la línea medioclavicular), por compromiso primario de la HCL o por hipertensión portal secundaria a cirrosis biliar, que a veces lleva a citopenias por hiperesplenismo y produce compromiso respiratorio. En general, la esplenectomía solo brinda alivio transitorio de las citopenias, porque el aumento del tamaño hepático y la activación reticuloendotelial causan secuestro y destrucción de los glóbulos sanguíneos en la sangre periférica. La esplenectomía solo se lleva a cabo como medida para salvar la vida del paciente.
Médula ósea
La mayoría de los pacientes con compromiso de la médula ósea son niños pequeños que tienen enfermedad difusa en el hígado, el bazo, los ganglios linfáticos y la piel, y que presentan trombocitopenia significativa (<100 000 × 109/l) y anemia (hemoglobina <10 g/dl; lactantes, <9 g/dl) que no son secundarias a otras causas, con leucopenia o sin esta (<4,0 × 109/l). Otros pacientes solo tienen citopenias leves y se observa compromiso de la médula ósea por la HCL que se identifica mediante inmunohistoquímica sensible, citometría de flujo o PCR para análisis de células con alteraciones en BRAF en la médula ósea. Un contenido alto de macrófagos a veces oculta las células de la HCL en la médula ósea. Los pacientes con HCL que se consideran de riesgo muy alto a veces presentan hemofagocitosis en la médula ósea. El entorno de citocinas que impulsa la HCL es probablemente responsable del epifenómeno de activación de macrófagos que, en los casos más graves, se presenta con manifestaciones típicas de linfohistiocitosis hemofagocítica, como citopenias e hiperferritinemia.
Tratamiento de la histiocitosis de células de Langerhans multisistémica de riesgo alto
Durante varios años, los grupos de estudio nacional e internacional han definido los grupos de tratamiento según el riesgo para asignar a los pacientes con histiocitosis de células de Langerhans (HCL) en función del riesgo de muerte y efectos tardíos por la enfermedad.
Según el sitio y la extensión de la enfermedad, es posible que el tratamiento de la HCL incluya observación (después de la biopsia o el curetaje), cirugía, radioterapia o fármacos por vía oral, tópica o intravenosa. La duración recomendada para el tratamiento es de 12 meses en los pacientes que necesitan quimioterapia cuando la enfermedad es monosistémica con compromiso óseo, cutáneo o ganglionar.
En los pacientes con enfermedad multisistémica de riesgo alto o bajo, la tasa de reactivación después de 6 meses de tratamiento fue de hasta el 50 % en los ensayos HISTSOC-LCH-I e HISTSOC-LCH-II. En los ensayos del grupo alemán-austriaco-neerlandés (DAL) se trataron a los pacientes durante 1 año y se presentaron menos recaídas (29 %). A partir de estos hallazgos, el ensayo HISTSOC-LCH-III se diseñó para administrar 12 meses de quimioterapia a todos los pacientes con enfermedad multisistémica de riesgo alto y para asignar al azar al tratamiento (6 o 12 meses) a los pacientes con enfermedad multisistémica de riesgo bajo. La tasa de reactivación se redujo significativamente hasta un 30 % en los pacientes con enfermedad de riesgo bajo o riesgo alto que recibieron 12 meses de tratamiento.
El tratamiento estándar para la HCL se basa en datos de ensayos internacionales con un gran número de pacientes. Sin embargo, algunos pacientes tienen HCL solo en la piel, la boca, la hipófisis u otros sitios que no se estudiaron en esos ensayos. En esos casos, las recomendaciones terapéuticas se fundamentan en series de casos que no cuentan con la solidez de la evidencia obtenida de los ensayos.
La Histiocyte Society organiza ensayos clínicos de tratamiento pediátrico en los que se han inscrito pacientes desde la década de 1980. La información en inglés sobre los centros que están inscribiendo pacientes en estos ensayos se encuentra en el portal de Internet ClinicalTrials.gov.
Las opciones de tratamiento para los pacientes de enfermedad multisistémica de riesgo alto (compromiso de bazo, hígado y médula ósea en uno o más sitios), son las siguientes:
- Quimioterapia.
Quimioterapia
Evidencia (quimioterapia):
- En los estudios HISTSOC-LCH-II e HISTSOC-LCH-III (NCT00276757), el grupo de tratamiento estándar consistió en vinblastina y prednisona, como se describió anteriormente, pero se añadió mercaptopurina a la fase de continuación del protocolo.[Nivel de evidencia A1]
- En la actualidad, la duración del tratamiento estándar para la HCL con compromiso del bazo, el hígado o la médula ósea (órganos de riesgo alto) es de 12 meses a partir de los estudios DAL-HX 83 y HISTSOC-LCH-III.
- En el estudio HISTSOC-LCH-II, los pacientes se asignaron al azar a tratamiento con vinblastina, prednisona y mercaptopurina o vinblastina, prednisona, mercaptopurina y etopósido.[Nivel de evidencia A1]
- No se observó ninguna diferencia estadísticamente significativa en los resultados (respuesta a las 6 semanas, probabilidad de supervivencia a 5 años, recaídas y consecuencias permanentes) entre los 2 grupos de tratamiento. En consecuencia, en los ensayos posteriores de la Histiocyte Society no se usó etopósido.
- Sin embargo, en una revisión posterior de los resultados, se notificó una reducción de la mortalidad en los pacientes del grupo de etopósido con compromiso de un órgano de riesgo.
- Aunque es controvertido, en una comparación de pacientes de los ensayos HISTSOC-LCH-I y HISTSOC-LCH-II se indicaron los siguientes resultados:
- El aumento de la intensidad del tratamiento generó otras respuestas tempranas y redujo la mortalidad.
- Cabe destacar que en esos estudios los pulmones se consideraron órganos de riesgo. No obstante, en análisis posteriores se observó que el compromiso pulmonar carece de importancia pronóstica.
- En el estudio HISTSOC-LCH-III (NCT00276757), los pacientes con compromiso en órganos de riesgo se asignaron al azar para recibir vinblastina, prednisona y mercaptopurina o vinblastina, prednisona y mercaptopurina con metotrexato (intravenoso en la fase de inducción y oral en la fase de continuación).
- Las tasas de respuesta a las 6 y 12 semanas y la SG no fueron diferentes entre los grupos; sin embargo, hubo un aumento significativo de los efectos tóxicos de grado 3 y 4 en los pacientes que recibieron metotrexato.
- Una conclusión importante del estudio HISTSOC-LCH-III fue que la supervivencia de los pacientes con HCL de riesgo alto en ambos grupos del estudio mejoró de manera significativa, en comparación con los pacientes del estudio anterior HISTSOC-LCH-II, aunque se usaron los mismos fármacos en el grupo estándar. Las siguientes son las posibles explicaciones para la mejora de la supervivencia:
- Una segunda fase de inducción de 6 semanas de vinblastina semanal con prednisona oral 3 días a la semana. Esta fase de reinducción se administró a todos los pacientes que no alcanzaron un estado de enfermedad inactiva al final de la fase de inducción de 6 semanas, antes de pasar a los cursos de mantenimiento cada 3 semanas. La tasa de la enfermedad inactiva aumentó después de la segunda fase de inducción; este curso quizás cumplió una función importante en la mejora de la tasa de supervivencia.
- Mejores cuidados médicos de apoyo.
- Cambio precoz a una estrategia de rescate eficaz para las lesiones que no respondieron al tratamiento.
- Cabe destacar que, aunque se mejoró la supervivencia en el estudio HISTSOC-LCH-III, solo el 60 % de los pacientes presentó enfermedad inactiva en los órganos de riesgo después de 1 año de tratamiento y del 25 % al 29 % de los pacientes recayó.
- En el ensayo JLSG-96 el tratamiento incluyó un régimen de inducción de 6 semanas con citarabina, vincristina y prednisolona, seguido de 6 meses de mantenimiento con citarabina, vincristina, prednisolona y dosis bajas de metotrexato intravenoso. Si los pacientes tenían una respuesta precaria al régimen inicial, se les cambió a un régimen de rescate de combinación intensiva de doxorrubicina, ciclofosfamida, metotrexato, vincristina y prednisolona.[Nivel de evidencia B1]
- La tasa de respuesta a 5 años fue del 78 %, y la tasa de SG fue del 95 % en los pacientes con enfermedad multisistémica.
- La diabetes insípida se presentó en el 8,9 % de los pacientes con enfermedad multisistémica.
- De forma similar al estudio HISTSOC-LCH-III (NCT00276757) un hallazgo importante en este estudio fue la disminución de la mortalidad, en comparación con estudios anteriores del JLSG y el estudio HISTSOC-LCH-II. Esto se atribuyó al cambio precoz a una terapia de rescate más eficaz para los pacientes sin respuesta al tratamiento y mejores cuidados médicos de apoyo.
- El estudio tuvo una tasa de reactivación alta, lo que provocó varios cambios, como un aumento de la duración del ensayo a 12 meses y la adición de vinblastina, prednisona, mercaptopurina y metotrexato.
- El protocolo JLSG-02 fue similar al del estudio JLSG-96, excepto que se añadió ciclosporina a la reinducción de pacientes con respuesta deficiente y la duración del tratamiento aumentó a 54 semanas para pacientes con respuesta adecuada y a 60 semanas para pacientes con respuesta deficiente.[Nivel de evidencia B1]
- A pesar de un aumento marcado de la intensidad del tratamiento, las tasas de supervivencia sin complicaciones (SSC) fueron de solo un 46 % en los pacientes de riesgo alto y de un 70 % en los pacientes de riesgo bajo. En el estudio HISTSOC-LCH-III, las tasas de SSC fueron del 33 % en los pacientes de riesgo alto y del 50 % en los pacientes de riesgo bajo.
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presentan ejemplos de ensayos clínicos nacionales o institucionales en curso:
- HISTSOC-LCH-IV (NCT02205762) (LCH-IV, International Collaborative Treatment Protocol for Children and Adolescents with LCH): a partir del cuadro clínico inicial y la respuesta al tratamiento, en el estudio LCH-IV se adapta el tratamiento de acuerdo con los 7 estratos que se describen a continuación:
- Estrato I. Tratamiento de primera línea para los pacientes con HCL multisistémica (grupo 1) y los pacientes con HCL monosistémica y lesiones óseas multifocales o lesiones de riesgo para el SNC (grupo 2).
- Estrato II. Tratamiento de segunda línea para los pacientes sin compromiso de órganos de riesgo (que no responden al tratamiento de primera línea o presentan una reactivación después de completar el tratamiento de primera línea).
- Estrato III. Tratamiento de rescate para los pacientes de HCL con compromiso de órganos de riesgo (pacientes con disfunción de órganos de riesgo que no responden al tratamiento de primera línea).
- Estrato IV. Trasplante de células madre hematopoyéticas para la HCL con compromiso de órganos de riesgo (pacientes con disfunción de órganos de riesgo que no reciben tratamiento de primera línea).
- Estrato V. Seguimiento y tratamiento de tumores aislados y HCL neurodegenerativa en el SNC.
- Estrato VI. Evolución natural y tratamiento de otro tipo de HCL monosistémica (pacientes que no necesitan tratamiento sistémico en el momento del diagnóstico).
- Estrato VII (seguimiento a largo plazo). Seguimiento de todos los pacientes, sin considerar el tratamiento previo, con el fin de vigilar la reactivación o las consecuencias permanentes una vez que se resuelve la enfermedad y se finaliza el protocolo de tratamiento.
- NCT02670707 (Cytarabine or Vinblastine Sulfate and Prednisone in Treating Patients With LCH): el propósito de este ensayo es comparar el uso de vinblastina y prednisona con la monoterapia de citarabina para el tratamiento de la HCL.
Es preferible que los pacientes con HCL se inscriban en un ensayo clínico en cuanto sea posible para lograr avances terapéuticos más pronto, aplicar las recomendaciones que se basan en la evidencia científica y asegurar una atención óptima. La información en inglés sobre los ensayos clínicos para niños con HCL se encuentra disponible en el portal de Internet del NCI, el portal de Internet de la Histiocyte Society y el portal de Internet del North American Consortium for Histiocytosis (NACHO).
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Histiocitosis de células de Langerhans infantil recidivante, resistente al tratamiento o progresiva
Reactivación de la histiocitosis de células de Langerhans monosistémica y multisistémica
Es común que se produzca la reactivación de la histiocitosis de células de Langerhans (HCL) después de una respuesta completa. En un estudio grande, el porcentaje de pacientes con reactivaciones fue del 9 % al 17,4 % para quienes tenían enfermedad de un solo sitio; del 37 %, enfermedad multifocal de un solo sistema; del 46 %, enfermedad multisistémica (sin compromiso de órganos de riesgo); y del 54 % cuando tenían compromiso de órganos de riesgo. El 43 % de las reactivaciones se presentaron en los huesos, el 11 % en los oídos, el 9 % en la piel y el 7 % presentaron diabetes insípida; un porcentaje más bajo de pacientes presentaron recaídas ganglionares, medulares y en órganos de riesgo. La mediana de tiempo hasta la reactivación fue de 12 a 15 meses en los pacientes con enfermedad de riesgo bajo y de 9 meses en los pacientes con enfermedad de riesgo alto. Un tercio de los pacientes presentó más de 1 reactivación, entre 9 y 14 meses después de la reactivación inicial. Los pacientes con reactivaciones tuvieron más probabilidades de presentar secuelas a largo plazo en los huesos, diabetes insípida u otros problemas endocrinos, óticos y pulmonares.
En una revisión integral de los ensayos clínicos del grupo alemán-austriaco-neerlandés (DAL) y la Histiocyte Society, se observó una tasa de reactivación del 46 % a los 5 años en pacientes con HCL multisistémica; la mayoría de las reactivaciones se produjeron dentro de los 2 años de la primera remisión. En el 44 % de los pacientes se presentó una segunda reactivación, de nuevo, dentro de los 2 años de la segunda remisión. El compromiso de los órganos de riesgo en estas reactivaciones se produjo solo en aquellos pacientes que inicialmente estaban en el grupo de riesgo alto (es decir, tenían compromiso del hígado, el bazo o la médula ósea en el momento del diagnóstico original).[Nivel de evidencia C2] La mayoría de las reactivaciones, incluso en pacientes con enfermedad de riesgo alto que respondieron al tratamiento inicial, se produjeron en huesos, piel u otras ubicaciones de riesgo bajo.
De acuerdo con estos hallazgos, el porcentaje de reactivaciones en la enfermedad multisistémica fue del 45 % en un ensayo realizado en Japón [Nivel de evidencia A1] y del 46 % en el ensayo HISTSOC-LCH-II. No hubo ninguna diferencia estadísticamente significativa en las reactivaciones entre los grupos de riesgo alto y riesgo bajo. En ambos estudios, el DAL-HX y el estudio japonés, se llegó a la conclusión de que el tratamiento intensificado aumentó la velocidad de la respuesta, en especial en niños pequeños y lactantes menores de 2 años. Esto, junto con el cambio rápido a la terapia de rescate para los que no obtuvieron respuestas favorables, redujo la mortalidad de los pacientes con HCL multisistémica de riesgo alto. Según el ensayo aleatorizado HISTSOC-LCH-III (NCT00276757), la prolongación del tratamiento también redujo significativamente la tasa de reactivación. En el ensayo HISTSOC-LCH-IV (NCT02205762) se aborda la duración óptima del tratamiento (12 vs. 24 meses).
Tratamiento de la histiocitosis de células de Langerhans monosistémica o multisistémica recidivante resistente al tratamiento o progresiva de riesgo bajo
Todavía no se ha determinado el tratamiento óptimo para los pacientes con histiocitosis de células de Langerhans (HCL) recidivante, resistente al tratamiento o progresiva.
Las opciones de tratamiento para los pacientes con HCL recidivante, resistente al tratamiento o progresiva, monosistémica o multisistémica, de riesgo bajo son las siguientes:
- Quimioterapia.
- Terapia con bisfosfonatos.
Quimioterapia
Los siguientes regímenes de quimioterapia se han usado para el tratamiento de pacientes con enfermedad recidivante, resistente al tratamiento o progresiva de riesgo bajo:
- Vinblastina y prednisona. Los pacientes con enfermedad ósea recidivante que reaparece meses después de interrumpir la vinblastina y la prednisona tal vez se beneficien del tratamiento de reinducción con vinblastina semanal y prednisona diaria por 6 meses. Si hay enfermedad inactiva o muy poca evidencia de enfermedad activa, el tratamiento se puede cambiar a cada 3 semanas, con mercaptopurina oral cada noche.
- Vincristina, prednisona y citarabina. En un régimen de tratamiento alternativo para pacientes con cualquier combinación de sitios de enfermedad de riesgo bajo se emplea vincristina, prednisona y citarabina.[Nivel de evidencia C3]
- Citarabina en monoterapia. La citarabina monoterapia en dosis de 100 a 170 mg/m2 por día durante 5 días también ha demostrado ser eficaz.
- Cladribina. La cladribina en dosis de 5 mg/m2 por día durante 5 días por ciclo ha demostrado eficacia para la HCL de riesgo bajo recidivante (HCL multifocal ósea y multisistémica de riesgo bajo), con muy poca toxicidad.[Nivel de evidencia C3] La terapia con cladribina se debe limitar, si es posible, a un máximo de 6 ciclos para evitar citopenias acumuladas y potencialmente duraderas.
En un estudio de 44 pacientes pediátricos con HCL de riesgo bajo que recibieron cladribina, 5 pacientes lograron una remisión completa después de una mediana de seguimiento de más de 5 años. Se presentó neutropenia de grado 3 o superior en el 32 % de los pacientes, y linfopenia de grado 3 o superior en el 72 % de los pacientes. Los pacientes con enfermedad estable o respuestas parciales después de 6 meses de tratamiento quizás logren una respuesta completa.
- Clofarabina. La clofarabina es un tratamiento eficaz probado para pacientes con recaídas múltiples de HCL de riesgo bajo o riesgo alto.[Nivel de evidencia C3]
- Hidroxiurea, sola o combinada con metotrexato oral. En un ensayo de un solo centro, el tratamiento con hidroxiurea, sola o en combinación con metotrexato oral, notificó los siguientes resultados:[Nivel de evidencia C3]
- De 15 pacientes con HCL recidivante de riesgo bajo, 12 tuvieron respuestas al tratamiento.
- Talidomida. En un ensayo de fase II de talidomida en pacientes de HCL (10 pacientes de riesgo bajo; 6 pacientes de riesgo alto) con fracaso del tratamiento primario y de, al menos, un régimen secundario, se demostró lo siguiente:[Nivel de evidencia C3]
- De los 10 pacientes de riesgo bajo, 4 lograron respuestas completas y 3 lograron respuestas parciales. La respuesta completa se definió como la curación de las lesiones óseas en las radiografías simples (n = 3) o la resolución completa del exantema (n = 4, incluso 3 pacientes con lesiones óseas presentaron resolución completa). La respuesta parcial se definió como la curación de una lesión ósea, pero seguida por un empeoramiento del exantema que se había resuelto de forma parcial.
- Los efectos tóxicos que limitan la dosis, como la neuropatía y la neutropenia, tal vez reduzcan la utilidad general de la talidomida.
- La talidomida no es un fármaco importante para el tratamiento de pacientes pediátricos.
Terapia con bisfosfonatos
La terapia con bisfosfonatos también es eficaz para el tratamiento de pacientes con lesiones óseas de HCL recidivantes.
Evidencia (bisfosfonatos):
- En una encuesta realizada en Japón, 16 pacientes con lesiones óseas recibieron bisfosfonatos. Ninguno tenía enfermedad en órganos de riesgo. La mayoría de los pacientes recibieron 6 ciclos de pamidronato de 1 mg/kg por curso, administrados a intervalos de 4 semanas.[Nivel de evidencia C3]
- De los 16 pacientes, 12 recibieron tratamiento exitoso.
- También se resolvieron las lesiones de la HCL en la piel y el tejido blando en los pacientes que tuvieron respuesta favorable.
- Al cabo de una mediana de 3,3 años, 8 de 12 pacientes permanecieron sin enfermedad.
- Otros bisfosfonatos, como el zoledronato y el alendronato oral, también han sido exitosos para el tratamiento de la HCL ósea.[Nivel de evidencia C3]
Tratamiento de la histiocitosis de células de Langerhans multisistémica recidivante, resistente al tratamiento o progresiva de riesgo alto
En los datos de los estudios del grupo DAL se observó que los pacientes con HCL multisistémica de riesgo alto que presentaron enfermedad progresiva antes de la semana 6 del tratamiento de inducción estándar, o que no obtuvieron por lo menos 1 respuesta parcial antes de la semana 12, solo tenían una probabilidad de supervivencia del 10 %. Estos resultados coincidieron con los del ensayo menos intensivo HISTSOC-LCH-II, en el que los pacientes tratados con vinblastina y prednisona que no respondieron bien hacia la semana 6 tuvieron una probabilidad de supervivencia del 27 %, en comparación con el 52 % en quienes respondieron bien.[Nivel de evidencia A1] Para mejorar estos resultados, los pacientes con respuesta deficiente deben pasar a estrategias de rescate hacia la semana 6 si presentan enfermedad progresiva y a más tardar hacia la semana 12 para aquellos que no presentan por lo menos una respuesta buena.
Las opciones de tratamiento para los pacientes con HCL multisistémica recidivante, resistente o progresiva de riesgo alto son las siguientes:
- Quimioterapia.
- Terapia dirigida (por ejemplo, inhibidores de MAPK).
- Trasplante de células madre hematopoyéticas.
Quimioterapia
Cladribina y citarabina
Evidencia (cladribina y citarabina):
- 10 pacientes con compromiso de órganos de riesgo alto (hígado, bazo o médula ósea) y compromiso multisistémico de órganos de riesgo bajo resistentes al tratamiento se trataron con un protocolo intensivo similar al usado para la leucemia mieloide aguda que incluía cladribina y citarabina.[Nivel de evidencia C3] En el ensayo de seguimiento HISTSOC-LCH-S-2005 participaron 27 pacientes y se observaron los siguientes resultados:
- La tasa de supervivencia sin progresión fue del 63 % y la tasa de SG a 5 años fue del 85 % en este grupo de pacientes de riesgo alto con resistencia al tratamiento.
- Todos los pacientes presentaron efectos tóxicos hematológicos de grado 4; además, se produjo septicemia grave en 5 de estos pacientes.
- Para los centros que no pueden proporcionar los cuidados intensivos necesarios para este protocolo, se publicó un protocolo alternativo con dosis bajas de cladribina (5 mg/m2/día × 5 días) y citarabina (100 mg/m2/día × 4 días).[Nivel de evidencia C2]
- De 9 pacientes, 6 alcanzaron el estado de enfermedad inactiva y 1 paciente mejoró el estado después de 6 ciclos.
- Algunos pacientes recibieron terapia de mantenimiento.
- Además, 7 de 9 pacientes permanecieron en remisión completa, al cabo de una mediana de seguimiento de 6,5 años.
Clofarabina
Se notificó que los pacientes que no respondieron al tratamiento con cladribina respondieron al tratamiento con clofarabina.; [Nivel de evidencia C2]
Evidencia (clofarabina):
- Un grupo de 11 pacientes con enfermedad multisistémica recidivante de riesgo alto y riesgo bajo se trataron con clofarabina.
- La tasa de SG fue del 90 %.
- Si se corroboran estos resultados en ensayos prospectivos, la toxicidad reducida de este régimen, en comparación con la combinación de cladribina y citarabina, ofrecería ventajas a pesar del costo del fármaco.
Terapia dirigida
Inhibidores de MAPK
El descubrimiento de que la mayoría de los pacientes con HCL presentan la variante BRAF V600E u otras variantes que activan la vía RAS indica que las terapias nuevas dirigidas a moléculas de esta vía (inhibidores de MAP2K/ERK) cumplirán una función importante en el tratamiento de la HCL.
Evidencia (vemurafenib):
- Se administró vemurafenib a 44 pacientes que tenían HCL con compromiso de órganos de riesgo y 10 pacientes sin compromiso de órganos de riesgo. De los 44 pacientes con compromiso de órganos de riesgo, 31 recibieron vemurafenib como tratamiento original y 13 recibieron vemurafenib después de la progresión de la enfermedad. Los 10 pacientes sin compromiso de órganos de riesgo también recibieron vemurafenib después de la progresión de la enfermedad.[Nivel de evidencia C3]
- Después de 8 semanas de tratamiento, 38 pacientes presentaron respuestas completas y 16 pacientes presentaron respuestas parciales. La mayoría de los pacientes se trataron durante 6 meses.
- En este estudio 30 pacientes suspendieron el vemurafenib; 24 de ellos recayeron después: El 72 % de los pacientes recayó a los 6 meses y el 84 % recayó a los 12 meses de suspender el tratamiento.
- La tasa de recaída fue del 95 % en los pacientes con compromiso de órganos de riesgo y del 57 % en los pacientes sin compromiso de órganos de riesgo.
- La recaída se relacionó con persistencia en la circulación de células positivas para BRAF.
- Los efectos adversos del fármaco más frecuentes fueron de tipo dermatológico. En una revisión de 57 pacientes con HCL que recibieron vemurafenib para la enfermedad resistente al tratamiento, el 72 % de los pacientes tuvo efectos adversos cutáneos, de los cuales el 86 % fueron de grado 1 o 2. La mayoría de los pacientes presentaron fotosensibilidad, queratosis pilar, erupciones maculares o foliculares, o xerosis. No se observaron tumores cutáneos.
- En una revisión sistemática y un metanálisis se evaluó la eficacia y la inocuidad del vemurafenib para el tratamiento de pacientes con HCL. Se evaluaron 416 estudios, 22 de los cuales cumplían con los criterios de inclusión. Hubo 104 pacientes con enfermedad en recaída o resistente al tratamiento y 3 pacientes con enfermedad recién diagnosticada.
- Con el tratamiento con vemurafenib, la mediana de tiempo hasta la primera respuesta fue de 1 semana y la mediana de tiempo hasta la mejor respuesta fue de 5,25 meses.
- De los pacientes. 62 (58 %) alcanzaron el estado de enfermedad inactiva y un 36 % presentaron disminución de la enfermedad activa.
- La tasa de respuesta general fue del 94,4 %.
- Entre los efectos tóxicos principales se incluyeron exantema y fotosensibilidad.
- Los autores concluyeron que el vemurafenib fue muy eficaz e inocuo para tratar a pacientes con HCL resistente al tratamiento, pero aún no se ha establecido la duración del tratamiento.
- En un análisis multicéntrico retrospectivo de experiencias en las que se usaron varios inhibidores de MAPK para tratar a 21 pacientes pediátricos con HCL con fracaso de, al menos, un tratamiento previo (mediana, 3 tratamientos previos), se demostró lo siguiente:[Nivel de evidencia C3]
- Tasa de respuesta general del 86 % (respuesta completa, 19 %; respuesta parcial, 67 %).
- Enfermedad estable en el 10 % de los pacientes.
- Los efectos tóxicos más frecuentes fueron erupciones cutáneas y artralgias. Otros efectos tóxicos fueron neutropenia, fatiga y uveitis.
Evidencia (dabrafenib con trametinib o sin este):
- En un estudio se comparó el dabrafenib solo (13 pacientes) con dabrafenib y trametinib (12 pacientes) para el tratamiento de pacientes con HCL en recaída o progresiva.
- Después de un seguimiento a 2 años, las respuestas fueron similares en ambos grupos (46,2 % de respuesta completa, 30,8 % de enfermedad regresiva y 23,1 % de enfermedad estable para el dabrafenib solo vs. 33,3 % de respuesta completa, 25,0 % de enfermedad regresiva y 25,0 % de enfermedad estable para la terapia combinada).
- Los efectos adversos también fueron similares e incluyeron pirexia y vómitos, tos y aumento de la creatinina sérica.
- En este estudio se indicaría que la terapia combinada no es más eficaz en pacientes con HCL. Sin embargo, se necesitan más datos.
Aunque se han notificado neoplasias malignas, como carcinoma de células escamosas, en adultos tratados con inhibidores de MAPK, no se han notificado dichas neoplasias en pacientes pediátricos. Al igual que los adultos, los niños desarrollan erupciones acneiformes, fotosensibilidad, diarrea y, a veces, mialgias.
Inhibidores de tirosina–cinasas
Evidencia (inhibidores de tirosina–cinasas):
- Se demostró que el imatinib disminuye la diferenciación de las células madre positivas para CD34 a células dendríticas. Se han publicado informes de casos pequeños sobre su eficacia en pacientes con HCL.
Trasplante de células madre hematopoyéticas
Se ha usado el trasplante de células madre hematopoyéticas (TCMH) en pacientes con compromiso multisistémico de órganos de riesgo alto resistente a la quimioterapia. En los resultados iniciales se observó una mortalidad relacionada con el tratamiento muy alta para los lactantes pequeños enfermos, lo que llevó al uso de un acondicionamiento de intensidad reducida.
Evidencia (acondicionamiento de intensidad reducida vs. acondicionamiento mielosupresor para el TCMH):
- En una revisión del Reino Unido se indicó que, en los centros de trasplantes que tienen experiencia en TCMH para la HCL, no se observaron ventajas de usar el acondicionamiento de intensidad reducida en este entorno.[Nivel de evidencia C2]
- El acondicionamiento de intensidad reducida no ofreció ninguna ventaja en relación con la SG en comparación con el acondicionamiento mielosupresor para los pacientes con HCL; la tasa de recaída después del acondicionamiento de intensidad reducida fue significativamente más alta (28 %) que la tasa de recaída después del acondicionamiento mielosupresor (8 %).
- Muchos de los pacientes que recibieron acondicionamiento de intensidad reducida y que presentaron recaídas se volvieron a tratar con éxito con quimioterapia sola.
Opciones de tratamiento para la colangitis esclerosante y la activación de macrófagos
De los niños con colangitis esclerosante, el 75 % no responderán a la quimioterapia porque la HCL está inactiva, aunque la fibrosis y la esclerosis permanecen. Pese a sus limitaciones, la biopsia de hígado tal vez sea la única forma de distinguir la HCL activa de la fibrosis terminal. Cuando empeora el funcionamiento hepático, el trasplante de hígado es la única opción de tratamiento. En una revisión de 60 pacientes con HCL (55 niños) sometidos a trasplante de hígado por insuficiencia hepática relacionada con HCL, se informó una tasa de supervivencia a 5 años del 82 %. Se produjo rechazo del trasplante en el 55 % de los pacientes y el 22 % de ellos recibió un segundo trasplante. La tasa de supervivencia general del injerto a 5 años fue del 62 % en los pacientes que recibieron un trasplante de donante fallecido y del 81 % en los pacientes que recibieron un trasplante de un donante vivo (no fue estadísticamente significativo). Murieron 9 pacientes (15 %). Hubo 1 caso de enfermedad linfoproliferativa postrasplante (TLPT). No hay datos de recidivas de HCL. Los autores llevaron a cabo una revisión bibliográfica para identificar otros 50 pacientes con HCL sometidos a un trasplante de hígado. Entre estos pacientes, el 47 % presentó rechazó, el 11 % TLPT y el 8 % HCL recidivante. Se trató a siete pacientes (14 %) con pérdida del injerto con un nuevo trasplante.[Nivel de evidencia C2]
En informes de casos y series de casos se ha documentado la eficacia de los inhibidores de MAPK para el tratamiento de la HCL hepática progresiva.
Algunos pacientes presentan activación de macrófagos en la médula. Esto resulta confuso para los médicos, que tal vez piensen que el paciente tiene una linfohistiocitosis hemofagocítica (LHH) y una HCL. No se sabe cuál es el mejor tratamiento de esta manifestación potencialmente mortal porque no suele responder bien al tratamiento estándar de la LHH. Se podría considerar la clofarabina, el anticuerpo anti-CD52 alemtuzumab y el trasplante de células madre alogénicas de intensidad reducida.[Nivel de evidencia C3] Se desconoce si las terapias más nuevas para LHH, como el anticuerpo contra el interferón-gamma o el inhibidor de JAK-STAT ruxolitinib, serán más eficaces para la activación de macrófagos en la HCL que las opciones anteriores.
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de un ensayo clínico nacional o institucional en curso:
- NCT04079179 (Cobimetinib for the Treatment of Refractory LCH): la inscripción en este estudio está abierta a niños o adultos con HCL en recaída o resistente al tratamiento o con otros trastornos histiocíticos recién diagnosticados, en recaída o resistentes al tratamiento.
Evaluación de la respuesta al tratamiento
La evaluación de la respuesta sigue siendo una de las áreas más difíciles del tratamiento de la HCL. Es más fácil cuando hay un área específica que se puede controlar por evaluación clínica o con ecografía, TC, TEP o IRM, como la piel, la hepatomegalia o la esplenomegalia y otras lesiones expansivas o líticas óseas. A pesar de esto, sigue siendo importante el análisis clínico, que incluye la evaluación del dolor y de otros síntomas.
Es posible que las lesiones óseas tarden muchos meses en sanar y es difícil evaluarlas con radiografías simples, aunque la esclerosis en la periferia de una lesión ósea indica curación. Las TC o IRM sirven para evaluar la respuesta de una masa de tejido blando que se relaciona con una lesión ósea, pero no son muy útiles para evaluar la respuesta de las lesiones líticas en los huesos. El resultado de las gammagrafías óseas con tecnecio Tc 99m seguirá dando positivo durante el proceso de curación de hueso. Las TEP a veces son útiles para controlar la respuesta al tratamiento porque la intensidad de la imagen de TEP disminuye con la respuesta de las lesiones y la curación ósea.
Para los niños y adultos con HCL pulmonar, la prueba de funcionamiento pulmonar y las TC de alta resolución son métodos sensibles para detectar la progresión de la enfermedad. Los cambios intersticiales residuales que reflejan fibrosis residual o quistes inactivos residuales se deben distinguir de la enfermedad activa; la gammagrafía con análogo de la somatostatina puede ser útil en este caso.
Opciones de tratamiento para la histiocitosis de células de Langerhans infantil que ya no se consideran eficaces
Los tratamientos que se han utilizado en el pasado, pero que ya no se recomiendan para la HCL infantil, incluyen ciclosporina e interferón α.
Tampoco se indican intervenciones quirúrgicas radicales. Cuando hay lesiones en la mandíbula, es posible que una cirugía extensa arruine la dentición secundaria. Se contraindica la resección quirúrgica de lesiones en la ingle o los genitales porque la quimioterapia puede curar estas lesiones.
El uso de radioterapia en la HCL se ha reducido bastante en los pacientes pediátricos, e incluso la radioterapia de dosis bajas se debe limitar a lesiones de un solo hueso, lesiones de cuerpo vertebral u otras lesiones de un solo hueso que comprimen la médula espinal o el nervio óptico y que no responden a la quimioterapia o que son dolorosas y no susceptibles de recibir otro tratamiento.
Enfermedad tardía y efectos del tratamiento de la histiocitosis de células de Langerhans infantil
La frecuencia general notificada de las consecuencias a largo plazo de la HCL oscila entre el 20 % y el 70 %. Los niños con compromiso de órganos de riesgo bajo (piel, huesos, ganglios linfáticos o hipófisis) tienen una probabilidad de cerca del 20 % de presentar secuelas a largo plazo.; [Nivel de evidencia B4] Los pacientes con compromiso multisistémico tienen una tasa notificada de complicaciones a largo plazo de cerca del 70 % cuando el tratamiento es de solo 6 meses. Sin embargo, no se ha informado del alcance de las secuelas a largo plazo en pacientes que reciben tratamiento durante un año.
La razón de esta amplia variación se debe a la definición de caso, el tamaño de la muestra, el tratamiento administrado, el método de recopilación de datos y la duración del seguimiento. En estos estudios de calidad de vida se informaron los siguientes resultados:
- En un estudio de calidad de vida en los sobrevivientes a largo plazo de HCL esquelética, no hubo ninguna diferencia significativa en los puntajes de calidad de vida, en comparación con los niños y adultos sanos del grupo de control. Además, los puntajes de calidad de vida fueron muy similares entre aquellos con secuelas permanentes y sin estas.
- En otro estudio de 40 pacientes a quienes se examinó de forma minuciosa para detectar efectos tardíos, se encontraron puntajes adversos de la calidad de vida en más del 50 % de los pacientes. De estos pacientes, un 75 % presentaron secuelas detectables a largo plazo. Los efectos secundarios más comunes fueron la disfunción hipotalámica o hipofisaria (50 %), la disfunción cognitiva (20 %) y el compromiso cerebeloso (17,5 %).
Los efectos tardíos de la HCL se presentan en las siguientes partes del cuerpo:
- Sistema endocrino. Los pacientes con diabetes insípida corren el riesgo de panhipopituitarismo y necesitan vigilancia minuciosa para que logren un crecimiento y desarrollo adecuados. En una revisión retrospectiva de 141 pacientes con HCL y diabetes insípida, el 43 % presentó deficiencia de la hormona del crecimiento (HC). El riesgo de deficiencia de la HC a 5 años en los niños con HCL y diabetes insípida fue del 35 %, y el riesgo a 10 años fue del 54 %. No se observó aumento de la reactivación de la HCL en los pacientes que recibieron HC, en comparación con quienes no la recibieron. Los problemas de crecimiento y desarrollo son más frecuentes debido a la edad temprana en el momento de la presentación y los efectos más tóxicos de la terapia de prednisona a largo plazo en niños muy pequeños.
- Órganos de los sentidos (hipoacusia). Se encontró hipoacusia en un 38 % de los niños tratados por HCL. El 70 % de los pacientes con HCL en este estudio presentaron compromiso auditivo, que incluyó secreción auricular, hinchazón de la mastoides e hipoacusia. De los pacientes con anomalías en la TC o la IRM en la mastoides, el 59 % tenían hipoacusia.[Nivel de evidencia C1]
- Sistema nervioso. Se informó sobre síntomas neurológicos secundarios a la compresión vertebral de las lesiones cervicales en 3 de 26 pacientes con HCL y lesiones de la columna vertebral. La HCL del SNC se presenta con mayor frecuencia en niños con HCL en la hipófisis o en los huesos de riesgo para el SNC (hueso mastoides, orbital o temporal). Algunos pacientes con lesiones craneales de riesgo para el SNC que logran una supervivencia prolongada quizás presenten alteraciones cognitivas importantes y anomalías en las IRM. Algunos pacientes presentan anomalías en el funcionamiento cerebeloso y en el comportamiento, mientras que otros presentan deficiencias sutiles en la memoria a corto plazo y en los potenciales evocados por el tronco encefálico.
- Esqueleto. En un 20 % de los pacientes se observan problemas ortopédicos por lesiones de la columna vertebral, el fémur, la tibia o el húmero. Estos problemas son el aplastamiento vertebral o la inestabilidad de la columna vertebral que puede causar escoliosis y asimetría facial o de extremidades.
- Aparato respiratorio. La enfermedad pulmonar difusa a veces da lugar a un funcionamiento pulmonar precario con mayor riesgo de infecciones y menos tolerancia al ejercicio. Estos pacientes se deben vigilar mediante pruebas de funcionamiento pulmonar, como la capacidad de difusión del monóxido de carbono y la relación entre el volumen residual y la capacidad pulmonar total.
- Aparato digestivo. La hepatopatía puede conducir a una colangitis esclerosante, que casi siempre es resistente a cualquier tratamiento diferente a un trasplante de hígado. Los problemas dentales caracterizados por la caída de dientes son importantes para algunos pacientes, por lo general, relacionados con una cirugía dental demasiado radical.
- Neoplasias subsiguientes. La insuficiencia de médula ósea secundaria a la HCL o al tratamiento es poco frecuente y se relaciona con un riesgo más alto de neoplasia maligna. Los pacientes con HCL tienen un riesgo más alto de lo normal de presentar cánceres secundarios.
Después del tratamiento se puede presentar leucemia (generalmente, leucemia mieloide aguda) y linfoma linfoblástico. En pocos pacientes se informó de una HCL acompañada de una neoplasia maligna simultánea; algunos de ellos primero presentaron la neoplasia maligna y luego apareció la HCL. Se notificaron 3 pacientes con leucemia linfoblástica aguda (LLA) de células T y HCL agresiva y, al igual que con todos los trastornos histiocíticos relacionados con neoplasias malignas linfoblásticas o posteriores, se encontraron los mismos cambios genéticos en ambas enfermedades, lo que indica un origen clonal común. En un estudio se notificaron 2 casos en los que se encontró clonalidad con el mismo genotipo de receptor de células T γ. Los autores de este estudio enfatizaron la plasticidad de los linfocitos que se convierten en células de Langerhans. En el segundo estudio se describió el caso de un paciente que presentó HCL después de una LLA de células T que tenía los mismos reordenamientos del gen del receptor de células T y variantes activadoras del gen NOTCH1.
En una publicación basada en una encuesta a los miembros de la Histiocyte Society y en una revisión bibliográfica, se notificaron 116 casos de pacientes afectados por un par de trastornos: HCL y neoplasia maligna infantil. Prevalecieron las leucemias y los trastornos mieloproliferativos (n = 58; 50,0 %) sobre los tumores sólidos (n = 43; 37,1 %) y los linfomas (n = 15; 12,9 %). En la mayoría de los niños, la neoplasia maligna fue posterior a la HCL (n = 69; 59,5 %). Sin embargo, en algunos casos se observó LLA, incluso LLA de células T, antes de la HCL o de una neoplasia histiocítica. Por lo general, el trastorno histiocítico presentó los mismos hallazgos genéticos subyacentes que la leucemia anterior.
En otro estudio se notificó un análisis poblacional de neoplasias malignas subsiguientes en pacientes pediátricos en la base de datos del Surveillance, Epidemiology, and End Results (SEER) Program desde 2000 hasta 2016. Entre 936 casos pediátricos, se identificaron 2 casos de linfoma no Hodgkin, 2 casos de linfoma de Hodgkin y 1 caso de LLA de células T. No obstante, la mediana de seguimiento fue de 38 meses, lo que quizás no sea suficiente para identificar tumores sólidos secundarios.
References
- Carstensen H, Ornvold K: The epidemiology of Langerhans cell histiocytosis in children in Denmark, 1975-89. [Abstract] Med Pediatr Oncol 21 (5): A-15, 387-8, 1993.
- Salotti JA, Nanduri V, Pearce MS, et al.: Incidence and clinical features of Langerhans cell histiocytosis in the UK and Ireland. Arch Dis Child 94 (5): 376-80, 2009.
- Stålemark H, Laurencikas E, Karis J, et al.: Incidence of Langerhans cell histiocytosis in children: a population-based study. Pediatr Blood Cancer 51 (1): 76-81, 2008.
- A multicentre retrospective survey of Langerhans' cell histiocytosis: 348 cases observed between 1983 and 1993. The French Langerhans' Cell Histiocytosis Study Group. Arch Dis Child 75 (1): 17-24, 1996.
- Guyot-Goubin A, Donadieu J, Barkaoui M, et al.: Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000-2004. Pediatr Blood Cancer 51 (1): 71-5, 2008.
- Liu H, Stiller CA, Crooks CJ, et al.: Incidence, prevalence and survival in patients with Langerhans cell histiocytosis: A national registry study from England, 2013-2019. Br J Haematol 199 (5): 728-738, 2022.
- Ribeiro KB, Degar B, Antoneli CB, et al.: Ethnicity, race, and socioeconomic status influence incidence of Langerhans cell histiocytosis. Pediatr Blood Cancer 62 (6): 982-7, 2015.
- Bhatia S, Nesbit ME, Egeler RM, et al.: Epidemiologic study of Langerhans cell histiocytosis in children. J Pediatr 130 (5): 774-84, 1997.
- Peckham-Gregory EC, Danysh HE, Brown AL, et al.: Evaluation of maternal and perinatal characteristics on childhood lymphoma risk: A population-based case-control study. Pediatr Blood Cancer 64 (5): , 2017.
- Peckham-Gregory EC, Chakraborty R, Scheurer ME, et al.: A genome-wide association study of LCH identifies a variant in SMAD6 associated with susceptibility. Blood 130 (20): 2229-2232, 2017.
- Venkatramani R, Rosenberg S, Indramohan G, et al.: An exploratory epidemiological study of Langerhans cell histiocytosis. Pediatr Blood Cancer 59 (7): 1324-6, 2012.
- Nicholson HS, Egeler RM, Nesbit ME: The epidemiology of Langerhans cell histiocytosis. Hematol Oncol Clin North Am 12 (2): 379-84, 1998.
- McClain K, Jin H, Gresik V, et al.: Langerhans cell histiocytosis: lack of a viral etiology. Am J Hematol 47 (1): 16-20, 1994.
- Jeziorski E, Senechal B, Molina TJ, et al.: Herpes-virus infection in patients with Langerhans cell histiocytosis: a case-controlled sero-epidemiological study, and in situ analysis. PLoS One 3 (9): e3262, 2008.
- Haupt R, Minkov M, Astigarraga I, et al.: Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer 60 (2): 175-84, 2013.
- Binkovitz LA, Olshefski RS, Adler BH: Coincidence FDG-PET in the evaluation of Langerhans' cell histiocytosis: preliminary findings. Pediatr Radiol 33 (9): 598-602, 2003.
- Phillips M, Allen C, Gerson P, et al.: Comparison of FDG-PET scans to conventional radiography and bone scans in management of Langerhans cell histiocytosis. Pediatr Blood Cancer 52 (1): 97-101, 2009.
- Ribeiro MJ, Idbaih A, Thomas C, et al.: 18F-FDG PET in neurodegenerative Langerhans cell histiocytosis : results and potential interest for an early diagnosis of the disease. J Neurol 255 (4): 575-80, 2008.
- Grois N, Prayer D, Prosch H, et al.: Course and clinical impact of magnetic resonance imaging findings in diabetes insipidus associated with Langerhans cell histiocytosis. Pediatr Blood Cancer 43 (1): 59-65, 2004.
- Ha SY, Helms P, Fletcher M, et al.: Lung involvement in Langerhans' cell histiocytosis: prevalence, clinical features, and outcome. Pediatrics 89 (3): 466-9, 1992.
- Prasad SR, Wang H, Rosas H, et al.: Fat-containing lesions of the liver: radiologic-pathologic correlation. Radiographics 25 (2): 321-31, 2005 Mar-Apr.
- Ferrell J, Sharp S, Kumar A, et al.: Discrepancies between F-18-FDG PET/CT findings and conventional imaging in Langerhans cell histiocytosis. Pediatr Blood Cancer 68 (4): e28891, 2021.
- Rameh V, Voss S, Bedoya MA, et al.: The added value of skeletal surveys in the initial evaluation of children diagnosed with Langerhans cell histiocytosis in the era of staging 18 F-FDG PET/CT: A retrospective study. Pediatr Blood Cancer 70 (1): e30057, 2023.
- Prayer D, Grois N, Prosch H, et al.: MR imaging presentation of intracranial disease associated with Langerhans cell histiocytosis. AJNR Am J Neuroradiol 25 (5): 880-91, 2004.
- Ronceray L, Pötschger U, Janka G, et al.: Pulmonary involvement in pediatric-onset multisystem Langerhans cell histiocytosis: effect on course and outcome. J Pediatr 161 (1): 129-33.e1-3, 2012.
- Gadner H, Grois N, Arico M, et al.: A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr 138 (5): 728-34, 2001.
- Gadner H, Grois N, Pötschger U, et al.: Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood 111 (5): 2556-62, 2008.
- Gadner H, Minkov M, Grois N, et al.: Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood 121 (25): 5006-14, 2013.
- Haupt R, Nanduri V, Calevo MG, et al.: Permanent consequences in Langerhans cell histiocytosis patients: a pilot study from the Histiocyte Society-Late Effects Study Group. Pediatr Blood Cancer 42 (5): 438-44, 2004.
- Minkov M, Prosch H, Steiner M, et al.: Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer 45 (6): 802-7, 2005.
- Héritier S, Emile JF, Barkaoui MA, et al.: BRAF Mutation Correlates With High-Risk Langerhans Cell Histiocytosis and Increased Resistance to First-Line Therapy. J Clin Oncol 34 (25): 3023-30, 2016.
- Berres ML, Lim KP, Peters T, et al.: BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 211 (4): 669-83, 2014.
- Kemps PG, Zondag TCE, Arnardóttir HB, et al.: Clinicogenomic associations in childhood Langerhans cell histiocytosis: an international cohort study. Blood Adv 7 (4): 664-679, 2023.
- Rodriguez-Galindo C, Allen CE: Langerhans cell histiocytosis. Blood 135 (16): 1319-1331, 2020.
- Wnorowski M, Prosch H, Prayer D, et al.: Pattern and course of neurodegeneration in Langerhans cell histiocytosis. J Pediatr 153 (1): 127-32, 2008.
- Yeh EA, Greenberg J, Abla O, et al.: Evaluation and treatment of Langerhans cell histiocytosis patients with central nervous system abnormalities: Current views and new vistas. Pediatr Blood Cancer 65 (1): , 2018.
- Allen CE, Flores R, Rauch R, et al.: Neurodegenerative central nervous system Langerhans cell histiocytosis and coincident hydrocephalus treated with vincristine/cytosine arabinoside. Pediatr Blood Cancer 54 (3): 416-23, 2010.
- Bernstrand C, Cederlund K, Henter JI: Pulmonary function testing and pulmonary Langerhans cell histiocytosis. Pediatr Blood Cancer 49 (3): 323-8, 2007.
- Nanduri VR, Pritchard J, Levitt G, et al.: Long term morbidity and health related quality of life after multi-system Langerhans cell histiocytosis. Eur J Cancer 42 (15): 2563-9, 2006.
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed December 15, 2023.
- Minkov M, Pötschger U, Thacker N, et al.: Additive Prognostic Impact of Gastrointestinal Involvement in Severe Multisystem Langerhans Cell Histiocytosis. J Pediatr 237: 65-70.e3, 2021.
- Donadieu J, Egeler RM, Pritchard J: Langerhans cell histiocytosis: a clinical update. In: Weitzman S, Egeler R M, eds.: Histiocytic Disorders of Children and Adults. Cambridge, United Kingdom: Cambridge University Press, 2005, pp 95-129.
- Slater JM, Swarm OJ: Eosinophilic granuloma of bone. Med Pediatr Oncol 8 (2): 151-64, 1980.
- Peng XS, Pan T, Chen LY, et al.: Langerhans' cell histiocytosis of the spine in children with soft tissue extension and chemotherapy. Int Orthop 33 (3): 731-6, 2009.
- Boztug K, Frimpong-Ansah K, Nanduri VR, et al.: Intraocular Langerhans cell histiocytosis in a neonate resulting in bilateral loss of vision. Pediatr Blood Cancer 47 (5): 633-5, 2006.
- Simko SJ, Garmezy B, Abhyankar H, et al.: Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr 165 (5): 990-6, 2014.
- Stein SL, Paller AS, Haut PR, et al.: Langerhans cell histiocytosis presenting in the neonatal period: a retrospective case series. Arch Pediatr Adolesc Med 155 (7): 778-83, 2001.
- Lau L, Krafchik B, Trebo MM, et al.: Cutaneous Langerhans cell histiocytosis in children under one year. Pediatr Blood Cancer 46 (1): 66-71, 2006.
- Munn S, Chu AC: Langerhans cell histiocytosis of the skin. Hematol Oncol Clin North Am 12 (2): 269-86, 1998.
- Hashimoto K, Griffin D, Kohsbaki M: Self-healing reticulohistiocytosis: a clinical, histologic, and ultrastructural study of the fourth case in the literature. Cancer 49 (2): 331-7, 1982.
- Ashena Z, Alavi S, Arzanian MT, et al.: Nail involvement in langerhans cell histiocytosis. Pediatr Hematol Oncol 24 (1): 45-51, 2007 Jan-Feb.
- Madrigal-Martínez-Pereda C, Guerrero-Rodríguez V, Guisado-Moya B, et al.: Langerhans cell histiocytosis: literature review and descriptive analysis of oral manifestations. Med Oral Patol Oral Cir Bucal 14 (5): E222-8, 2009.
- Hicks J, Flaitz CM: Langerhans cell histiocytosis: current insights in a molecular age with emphasis on clinical oral and maxillofacial pathology practice. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 100 (2 Suppl): S42-66, 2005.
- Ducassou S, Seyrig F, Thomas C, et al.: Thymus and mediastinal node involvement in childhood Langerhans cell histiocytosis: long-term follow-up from the French national cohort. Pediatr Blood Cancer 60 (11): 1759-65, 2013.
- Vassallo R, Ryu JH, Colby TV, et al.: Pulmonary Langerhans'-cell histiocytosis. N Engl J Med 342 (26): 1969-78, 2000.
- Le Louet S, Barkaoui MA, Miron J, et al.: Childhood Langerhans cell histiocytosis with severe lung involvement: a nationwide cohort study. Orphanet J Rare Dis 15 (1): 241, 2020.
- Odame I, Li P, Lau L, et al.: Pulmonary Langerhans cell histiocytosis: a variable disease in childhood. Pediatr Blood Cancer 47 (7): 889-93, 2006.
- Abbritti M, Mazzei MA, Bargagli E, et al.: Utility of spiral CAT scan in the follow-up of patients with pulmonary Langerhans cell histiocytosis. Eur J Radiol 81 (8): 1907-12, 2012.
- Burnett A, Carney D, Mukhopadhyay S, et al.: Thyroid involvement with Langerhans cell histiocytosis in a 3-year-old male. Pediatr Blood Cancer 50 (3): 726-7, 2008.
- Imashuku S, Kinugawa N, Matsuzaki A, et al.: Langerhans cell histiocytosis with multifocal bone lesions: comparative clinical features between single and multi-systems. Int J Hematol 90 (4): 506-12, 2009.
- Aricò M, Astigarraga I, Braier J, et al.: Lack of bone lesions at diagnosis is associated with inferior outcome in multisystem langerhans cell histiocytosis of childhood. Br J Haematol 169 (2): 241-8, 2015.
- Goyal R, Das A, Nijhawan R, et al.: Langerhans cell histiocytosis infiltration into pancreas and kidney. Pediatr Blood Cancer 49 (5): 748-50, 2007.
- Hait E, Liang M, Degar B, et al.: Gastrointestinal tract involvement in Langerhans cell histiocytosis: case report and literature review. Pediatrics 118 (5): e1593-9, 2006.
- Geissmann F, Thomas C, Emile JF, et al.: Digestive tract involvement in Langerhans cell histiocytosis. The French Langerhans Cell Histiocytosis Study Group. J Pediatr 129 (6): 836-45, 1996.
- Donadieu J, Rolon MA, Thomas C, et al.: Endocrine involvement in pediatric-onset Langerhans' cell histiocytosis: a population-based study. J Pediatr 144 (3): 344-50, 2004.
- Robison NJ, Prabhu SP, Sun P, et al.: Predictors of neoplastic disease in children with isolated pituitary stalk thickening. Pediatr Blood Cancer 60 (10): 1630-5, 2013.
- Prosch H, Grois N, Prayer D, et al.: Central diabetes insipidus as presenting symptom of Langerhans cell histiocytosis. Pediatr Blood Cancer 43 (5): 594-9, 2004.
- Richards GE, Thomsett MJ, Boston BA, et al.: Natural history of idiopathic diabetes insipidus. J Pediatr 159 (4): 566-70, 2011.
- Di Iorgi N, Allegri AE, Napoli F, et al.: Central diabetes insipidus in children and young adults: etiological diagnosis and long-term outcome of idiopathic cases. J Clin Endocrinol Metab 99 (4): 1264-72, 2014.
- Marchand I, Barkaoui MA, Garel C, et al.: Central diabetes insipidus as the inaugural manifestation of Langerhans cell histiocytosis: natural history and medical evaluation of 26 children and adolescents. J Clin Endocrinol Metab 96 (9): E1352-60, 2011.
- Grois N, Pötschger U, Prosch H, et al.: Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr Blood Cancer 46 (2): 228-33, 2006.
- Shioda Y, Adachi S, Imashuku S, et al.: Analysis of 43 cases of Langerhans cell histiocytosis (LCH)-induced central diabetes insipidus registered in the JLSG-96 and JLSG-02 studies in Japan. Int J Hematol 94 (6): 545-51, 2011.
- Sakamoto K, Morimoto A, Shioda Y, et al.: Central diabetes insipidus in pediatric patients with Langerhans cell histiocytosis: Results from the JLSG-96/02 studies. Pediatr Blood Cancer 66 (1): e27454, 2019.
- Gadner H, Heitger A, Grois N, et al.: Treatment strategy for disseminated Langerhans cell histiocytosis. DAL HX-83 Study Group. Med Pediatr Oncol 23 (2): 72-80, 1994.
- Dunger DB, Broadbent V, Yeoman E, et al.: The frequency and natural history of diabetes insipidus in children with Langerhans-cell histiocytosis. N Engl J Med 321 (17): 1157-62, 1989.
- Grois NG, Favara BE, Mostbeck GH, et al.: Central nervous system disease in Langerhans cell histiocytosis. Hematol Oncol Clin North Am 12 (2): 287-305, 1998.
- Grois N, Prayer D, Prosch H, et al.: Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain 128 (Pt 4): 829-38, 2005.
- Fahrner B, Prosch H, Minkov M, et al.: Long-term outcome of hypothalamic pituitary tumors in Langerhans cell histiocytosis. Pediatr Blood Cancer 58 (4): 606-10, 2012.
- Héritier S, Barkaoui MA, Miron J, et al.: Incidence and risk factors for clinical neurodegenerative Langerhans cell histiocytosis: a longitudinal cohort study. Br J Haematol 183 (4): 608-617, 2018.
- McClain KL, Picarsic J, Chakraborty R, et al.: CNS Langerhans cell histiocytosis: Common hematopoietic origin for LCH-associated neurodegeneration and mass lesions. Cancer 124 (12): 2607-2620, 2018.
- Mittheisz E, Seidl R, Prayer D, et al.: Central nervous system-related permanent consequences in patients with Langerhans cell histiocytosis. Pediatr Blood Cancer 48 (1): 50-6, 2007.
- Minkov M, Steiner M, Pötschger U, et al.: Reactivations in multisystem Langerhans cell histiocytosis: data of the international LCH registry. J Pediatr 153 (5): 700-5, 705.e1-2, 2008.
- Braier JL, Rosso D, Latella A, et al.: Importance of multi-lineage hematologic involvement and hypoalbuminemia at diagnosis in patients with "risk-organ" multi-system Langerhans cell histiocytosis. J Pediatr Hematol Oncol 32 (4): e122-5, 2010.
- Steen AE, Steen KH, Bauer R, et al.: Successful treatment of cutaneous Langerhans cell histiocytosis with low-dose methotrexate. Br J Dermatol 145 (1): 137-40, 2001.
- Zinn DJ, Grimes AB, Lin H, et al.: Hydroxyurea: a new old therapy for Langerhans cell histiocytosis. Blood 128 (20): 2462-2465, 2016.
- McClain KL, Kozinetz CA: A phase II trial using thalidomide for Langerhans cell histiocytosis. Pediatr Blood Cancer 48 (1): 44-9, 2007.
- Uppuluri R, Ramachandrakurup S, Subburaj D, et al.: Excellent remission rates with limited toxicity in relapsed/refractory Langerhans cell histiocytosis with pulse dexamethasone and lenalidomide in children. Pediatr Blood Cancer 64 (1): 110-112, 2017.
- Hoeger PH, Nanduri VR, Harper JI, et al.: Long term follow up of topical mustine treatment for cutaneous langerhans cell histiocytosis. Arch Dis Child 82 (6): 483-7, 2000.
- Lindahl LM, Fenger-Grøn M, Iversen L: Topical nitrogen mustard therapy in patients with Langerhans cell histiocytosis. Br J Dermatol 166 (3): 642-5, 2012.
- Kwon OS, Cho KH, Song KY: Primary cutaneous Langerhans cell histiocytosis treated with photochemotherapy. J Dermatol 24 (1): 54-6, 1997.
- Vogel CA, Aughenbaugh W, Sharata H: Excimer laser as adjuvant therapy for adult cutaneous Langerhans cell histiocytosis. Arch Dermatol 144 (10): 1287-90, 2008.
- Laird J, Ma J, Chau K, et al.: Outcome After Radiation Therapy for Langerhans Cell Histiocytosis Is Dependent on Site of Involvement. Int J Radiat Oncol Biol Phys 100 (3): 670-678, 2018.
- Selch MT, Parker RG: Radiation therapy in the management of Langerhans cell histiocytosis. Med Pediatr Oncol 18 (2): 97-102, 1990.
- Kotecha R, Venkatramani R, Jubran RF, et al.: Clinical outcomes of radiation therapy in the management of Langerhans cell histiocytosis. Am J Clin Oncol 37 (6): 592-6, 2014.
- Greenberger JS, Cassady JR, Jaffe N, et al.: Radiation therapy in patients with histiocytosis: management of diabetes insipidus and bone lesions. Int J Radiat Oncol Biol Phys 5 (10): 1749-55, 1979.
- Nauert C, Zornoza J, Ayala A, et al.: Eosinophilic granuloma of bone: diagnosis and management. Skeletal Radiol 10 (4): 227-35, 1983.
- Gramatovici R, D'Angio GJ: Radiation therapy in soft-tissue lesions in histiocytosis X (Langerhans' cell histiocytosis). Med Pediatr Oncol 16 (4): 259-62, 1988.
- Baptista AM, Camargo AF, de Camargo OP, et al.: Does adjunctive chemotherapy reduce remission rates compared to cortisone alone in unifocal or multifocal histiocytosis of bone? Clin Orthop Relat Res 470 (3): 663-9, 2012.
- Willis B, Ablin A, Weinberg V, et al.: Disease course and late sequelae of Langerhans' cell histiocytosis: 25-year experience at the University of California, San Francisco. J Clin Oncol 14 (7): 2073-82, 1996.
- Woo KI, Harris GJ: Eosinophilic granuloma of the orbit: understanding the paradox of aggressive destruction responsive to minimal intervention. Ophthal Plast Reconstr Surg 19 (6): 429-39, 2003.
- Gatineau-Sailliant S, Grimard P, Miron MC, et al.: Langerhans Cell Histiocytosis With Vertebral Involvement Diagnosed and Treated Over the Last 15 Years in a Single Canadian Pediatric Academic Institution. J Pediatr Hematol Oncol 42 (3): 222-227, 2020.
- Lau LM, Stuurman K, Weitzman S: Skeletal Langerhans cell histiocytosis in children: permanent consequences and health-related quality of life in long-term survivors. Pediatr Blood Cancer 50 (3): 607-12, 2008.
- Mammano S, Candiotto S, Balsano M: Cast and brace treatment of eosinophilic granuloma of the spine: long-term follow-up. J Pediatr Orthop 17 (6): 821-7, 1997 Nov-Dec.
- Titgemeyer C, Grois N, Minkov M, et al.: Pattern and course of single-system disease in Langerhans cell histiocytosis data from the DAL-HX 83- and 90-study. Med Pediatr Oncol 37 (2): 108-14, 2001.
- Morimoto A, Shioda Y, Imamura T, et al.: Intensification of induction therapy and prolongation of maintenance therapy did not improve the outcome of pediatric Langerhans cell histiocytosis with single-system multifocal bone lesions: results of the Japan Langerhans Cell Histiocytosis Study Group-02 Protocol Study. Int J Hematol 108 (2): 192-198, 2018.
- Egeler RM, de Kraker J, Voûte PA: Cytosine-arabinoside, vincristine, and prednisolone in the treatment of children with disseminated Langerhans cell histiocytosis with organ dysfunction: experience at a single institution. Med Pediatr Oncol 21 (4): 265-70, 1993.
- Weitzman S, Braier J, Donadieu J, et al.: 2'-Chlorodeoxyadenosine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH). results of the LCH-S-98 protocol of the Histiocyte Society. Pediatr Blood Cancer 53 (7): 1271-6, 2009.
- Farran RP, Zaretski E, Egeler RM: Treatment of Langerhans cell histiocytosis with pamidronate. J Pediatr Hematol Oncol 23 (1): 54-6, 2001.
- Chellapandian D, Makras P, Kaltsas G, et al.: Bisphosphonates in Langerhans Cell Histiocytosis: An International Retrospective Case Series. Mediterr J Hematol Infect Dis 8 (1): e2016033, 2016.
- Morimoto A, Shioda Y, Imamura T, et al.: Nationwide survey of bisphosphonate therapy for children with reactivated Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer 56 (1): 110-5, 2011.
- Sivendran S, Harvey H, Lipton A, et al.: Treatment of Langerhans cell histiocytosis bone lesions with zoledronic acid: a case series. Int J Hematol 93 (6): 782-6, 2011.
- Büchler T, Cervinek L, Belohlavek O, et al.: Langerhans cell histiocytosis with central nervous system involvement: follow-up by FDG-PET during treatment with cladribine. Pediatr Blood Cancer 44 (3): 286-8, 2005.
- Watts J, Files B: Langerhans cell histiocytosis: central nervous system involvement treated successfully with 2-chlorodeoxyadenosine. Pediatr Hematol Oncol 18 (3): 199-204, 2001 Apr-May.
- Dhall G, Finlay JL, Dunkel IJ, et al.: Analysis of outcome for patients with mass lesions of the central nervous system due to Langerhans cell histiocytosis treated with 2-chlorodeoxyadenosine. Pediatr Blood Cancer 50 (1): 72-9, 2008.
- Grois N, Fahrner B, Arceci RJ, et al.: Central nervous system disease in Langerhans cell histiocytosis. J Pediatr 156 (6): 873-81, 881.e1, 2010.
- Ng Wing Tin S, Martin-Duverneuil N, Idbaih A, et al.: Efficacy of vinblastine in central nervous system Langerhans cell histiocytosis: a nationwide retrospective study. Orphanet J Rare Dis 6 (1): 83, 2011.
- Imashuku S, Arceci RJ: Strategies for the Prevention of Central Nervous System Complications in Patients with Langerhans Cell Histiocytosis: The Problem of Neurodegenerative Syndrome. Hematol Oncol Clin North Am 29 (5): 875-93, 2015.
- Idbaih A, Donadieu J, Barthez MA, et al.: Retinoic acid therapy in "degenerative-like" neuro-langerhans cell histiocytosis: a prospective pilot study. Pediatr Blood Cancer 43 (1): 55-8, 2004.
- Imashuku S, Ishida S, Koike K, et al.: Cerebellar ataxia in pediatric patients with Langerhans cell histiocytosis. J Pediatr Hematol Oncol 26 (11): 735-9, 2004.
- Imashuku S, Okazaki NA, Nakayama M, et al.: Treatment of neurodegenerative CNS disease in Langerhans cell histiocytosis with a combination of intravenous immunoglobulin and chemotherapy. Pediatr Blood Cancer 50 (2): 308-11, 2008.
- Chohan G, Barnett Y, Gibson J, et al.: Langerhans cell histiocytosis with refractory central nervous system involvement responsive to infliximab. J Neurol Neurosurg Psychiatry 83 (5): 573-5, 2012.
- Eckstein OS, Visser J, Rodriguez-Galindo C, et al.: Clinical responses and persistent BRAF V600E+ blood cells in children with LCH treated with MAPK pathway inhibition. Blood 133 (15): 1691-1694, 2019.
- Morimoto A, Ikushima S, Kinugawa N, et al.: Improved outcome in the treatment of pediatric multifocal Langerhans cell histiocytosis: Results from the Japan Langerhans Cell Histiocytosis Study Group-96 protocol study. Cancer 107 (3): 613-9, 2006.
- Eckstein O, McAtee CL, Greenberg J, et al.: Rituximab therapy for patients with Langerhans cell histiocytosis-associated neurologic dysfunction. Pediatr Hematol Oncol 35 (7-8): 427-433, 2018 Oct - Nov.
- Wong A, Ortiz-Neira CL, Reslan WA, et al.: Liver involvement in Langerhans cell histiocytosis. Pediatr Radiol 36 (10): 1105-7, 2006.
- Jaffe R: Liver involvement in the histiocytic disorders of childhood. Pediatr Dev Pathol 7 (3): 214-25, 2004 May-Jun.
- Braier J, Ciocca M, Latella A, et al.: Cholestasis, sclerosing cholangitis, and liver transplantation in Langerhans cell Histiocytosis. Med Pediatr Oncol 38 (3): 178-82, 2002.
- Carrere X, Pinto N, Gene Olaciregui N, et al.: High prevalence of BRAFV600E in patients with cholestasis, sclerosing cholangitis or liver fibrosis secondary to Langerhans cell histiocytosis. Pediatr Blood Cancer 68 (7): e29115, 2021.
- McClain K, Ramsay NK, Robison L, et al.: Bone marrow involvement in histiocytosis X. Med Pediatr Oncol 11 (3): 167-71, 1983.
- Minkov M, Pötschger U, Grois N, et al.: Bone marrow assessment in Langerhans cell histiocytosis. Pediatr Blood Cancer 49 (5): 694-8, 2007.
- Ballester LY, Cantu MD, Lim KPH, et al.: The use of BRAF V600E mutation-specific immunohistochemistry in pediatric Langerhans cell histiocytosis. Hematol Oncol 36 (1): 307-315, 2018.
- Galluzzo ML, Braier J, Rosenzweig SD, et al.: Bone marrow findings at diagnosis in patients with multisystem langerhans cell histiocytosis. Pediatr Dev Pathol 13 (2): 101-6, 2010 Mar-Apr.
- Favara BE, Jaffe R, Egeler RM: Macrophage activation and hemophagocytic syndrome in langerhans cell histiocytosis: report of 30 cases. Pediatr Dev Pathol 5 (2): 130-40, 2002 Mar-Apr.
- Morimoto A, Shioda Y, Imamura T, et al.: Intensified and prolonged therapy comprising cytarabine, vincristine and prednisolone improves outcome in patients with multisystem Langerhans cell histiocytosis: results of the Japan Langerhans Cell Histiocytosis Study Group-02 Protocol Study. Int J Hematol 104 (1): 99-109, 2016.
- Allen CE, Merad M, McClain KL: Langerhans-Cell Histiocytosis. N Engl J Med 379 (9): 856-868, 2018.
- Pollono D, Rey G, Latella A, et al.: Reactivation and risk of sequelae in Langerhans cell histiocytosis. Pediatr Blood Cancer 48 (7): 696-9, 2007.
- Simko SJ, McClain KL, Allen CE: Up-front therapy for LCH: is it time to test an alternative to vinblastine/prednisone? Br J Haematol 169 (2): 299-301, 2015.
- Barkaoui MA, Queheille E, Aladjidi N, et al.: Long-term follow-up of children with risk organ-negative Langerhans cell histiocytosis after 2-chlorodeoxyadenosine treatment. Br J Haematol 191 (5): 825-834, 2020.
- Simko SJ, Tran HD, Jones J, et al.: Clofarabine salvage therapy in refractory multifocal histiocytic disorders, including Langerhans cell histiocytosis, juvenile xanthogranuloma and Rosai-Dorfman disease. Pediatr Blood Cancer 61 (3): 479-87, 2014.
- Kudo K, Ohga S, Morimoto A, et al.: Improved outcome of refractory Langerhans cell histiocytosis in children with hematopoietic stem cell transplantation in Japan. Bone Marrow Transplant 45 (5): 901-6, 2010.
- Imamura T, Sato T, Shiota Y, et al.: Outcome of pediatric patients with Langerhans cell histiocytosis treated with 2 chlorodeoxyadenosine: a nationwide survey in Japan. Int J Hematol 91 (4): 646-51, 2010.
- Donadieu J, Bernard F, van Noesel M, et al.: Cladribine and cytarabine in refractory multisystem Langerhans cell histiocytosis: results of an international phase 2 study. Blood 126 (12): 1415-23, 2015.
- Rosso DA, Amaral D, Latella A, et al.: Reduced doses of cladribine and cytarabine regimen was effective and well tolerated in patients with refractory-risk multisystem Langerhans cell histiocytosis. Br J Haematol 172 (2): 287-90, 2016.
- Rodriguez-Galindo C, Jeng M, Khuu P, et al.: Clofarabine in refractory Langerhans cell histiocytosis. Pediatr Blood Cancer 51 (5): 703-6, 2008.
- Abraham A, Alsultan A, Jeng M, et al.: Clofarabine salvage therapy for refractory high-risk langerhans cell histiocytosis. Pediatr Blood Cancer 60 (6): E19-22, 2013.
- Donadieu J, Larabi IA, Tardieu M, et al.: Vemurafenib for Refractory Multisystem Langerhans Cell Histiocytosis in Children: An International Observational Study. J Clin Oncol 37 (31): 2857-2865, 2019.
- Tardieu M, Néron A, Duvert-Lehembre S, et al.: Cutaneous adverse events in children treated with vemurafenib for refractory BRAFV600E mutated Langerhans cell histiocytosis. Pediatr Blood Cancer 68 (9): e29140, 2021.
- Mohapatra D, Gupta AK, Haldar P, et al.: Efficacy and safety of vemurafenib in Langerhans cell histiocytosis (LCH): A systematic review and meta-analysis. Pediatr Hematol Oncol 40 (1): 86-97, 2023.
- Whitlock JA, Geoerger B, Dunkel IJ, et al.: Dabrafenib, alone or in combination with trametinib, in BRAF V600-mutated pediatric Langerhans cell histiocytosis. Blood Adv 7 (15): 3806-3815, 2023.
- Janku F, Amin HM, Yang D, et al.: Response of histiocytoses to imatinib mesylate: fire to ashes. J Clin Oncol 28 (31): e633-6, 2010.
- Wagner C, Mohme H, Krömer-Olbrisch T, et al.: Langerhans cell histiocytosis: treatment failure with imatinib. Arch Dermatol 145 (8): 949-50, 2009.
- Akkari V, Donadieu J, Piguet C, et al.: Hematopoietic stem cell transplantation in patients with severe Langerhans cell histiocytosis and hematological dysfunction: experience of the French Langerhans Cell Study Group. Bone Marrow Transplant 31 (12): 1097-103, 2003.
- Nagarajan R, Neglia J, Ramsay N, et al.: Successful treatment of refractory Langerhans cell histiocytosis with unrelated cord blood transplantation. J Pediatr Hematol Oncol 23 (9): 629-32, 2001.
- Caselli D, Aricò M; EBMT Paediatric Working Party: The role of BMT in childhood histiocytoses. Bone Marrow Transplant 41 (Suppl 2): S8-S13, 2008.
- Kudo K, Maeda M, Suzuki N, et al.: Nationwide retrospective review of hematopoietic stem cell transplantation in children with refractory Langerhans cell histiocytosis. Int J Hematol 111 (1): 137-148, 2020.
- Veys PA, Nanduri V, Baker KS, et al.: Haematopoietic stem cell transplantation for refractory Langerhans cell histiocytosis: outcome by intensity of conditioning. Br J Haematol 169 (5): 711-8, 2015.
- Ziogas IA, Kakos CD, Wu WK, et al.: Liver Transplantation for Langerhans Cell Histiocytosis: A US Population-Based Analysis and Systematic Review of the Literature. Liver Transpl 27 (8): 1181-1190, 2021.
- Lee LH, Krupski C, Clark J, et al.: High-risk LCH in infants is serially transplantable in a xenograft model but responds durably to targeted therapy. Blood Adv 4 (4): 717-727, 2020.
- Jordan MB, McClain KL, Yan X, et al.: Anti-CD52 antibody, alemtuzumab, binds to Langerhans cells in Langerhans cell histiocytosis. Pediatr Blood Cancer 44 (3): 251-4, 2005.
- Tazi A, Hiltermann J, Vassallo R: Adult lung histiocytosis. In: Weitzman S, Egeler R M, eds.: Histiocytic Disorders of Children and Adults. Cambridge, United Kingdom: Cambridge University Press, 2005, pp 187-207.
- Minkov M, Grois N, Broadbent V, et al.: Cyclosporine A therapy for multisystem langerhans cell histiocytosis. Med Pediatr Oncol 33 (5): 482-5, 1999.
- Lukina EA, Kuznetsov VP, Beliaev DL, et al.: [The treatment of histiocytosis X (Langerhans-cell histiocytosis) with alpha-interferon preparations] Ter Arkh 65 (11): 67-70, 1993.
- Gadner H, Ladisch S: The treatment of Langerhans cell histiocytosis. In: Weitzman S, Egeler R M, eds.: Histiocytic Disorders of Children and Adults. Cambridge, United Kingdom: Cambridge University Press, 2005, pp 229-53.
- Chow TW, Leung WK, Cheng FWT, et al.: Late outcomes in children with Langerhans cell histiocytosis. Arch Dis Child 102 (9): 830-835, 2017.
- Sakamoto K, Morimoto A, Shioda Y, et al.: Long-term complications in uniformly treated paediatric Langerhans histiocytosis patients disclosed by 12 years of follow-up of the JLSG-96/02 studies. Br J Haematol 192 (3): 615-620, 2021.
- Donadieu J, Rolon MA, Pion I, et al.: Incidence of growth hormone deficiency in pediatric-onset Langerhans cell histiocytosis: efficacy and safety of growth hormone treatment. J Clin Endocrinol Metab 89 (2): 604-9, 2004.
- Komp DM: Long-term sequelae of histiocytosis X. Am J Pediatr Hematol Oncol 3 (2): 163-8, 1981.
- Nanduri V, Tatevossian R, Sirimanna T: High incidence of hearing loss in long-term survivors of multisystem Langerhans cell histiocytosis. Pediatr Blood Cancer 54 (3): 449-53, 2010.
- Nanduri VR, Lillywhite L, Chapman C, et al.: Cognitive outcome of long-term survivors of multisystem langerhans cell histiocytosis: a single-institution, cross-sectional study. J Clin Oncol 21 (15): 2961-7, 2003.
- Guimarães LF, Dias PF, Janini ME, et al.: Langerhans cell histiocytosis: impact on the permanent dentition after an 8-year follow-up. J Dent Child (Chic) 75 (1): 64-8, 2008 Jan-Apr.
- Egeler RM, Neglia JP, Puccetti DM, et al.: Association of Langerhans cell histiocytosis with malignant neoplasms. Cancer 71 (3): 865-73, 1993.
- Egeler RM, Neglia JP, Aricò M, et al.: The relation of Langerhans cell histiocytosis to acute leukemia, lymphomas, and other solid tumors. The LCH-Malignancy Study Group of the Histiocyte Society. Hematol Oncol Clin North Am 12 (2): 369-78, 1998.
- Castro EC, Blazquez C, Boyd J, et al.: Clinicopathologic features of histiocytic lesions following ALL, with a review of the literature. Pediatr Dev Pathol 13 (3): 225-37, 2010 May-Jun.
- Feldman AL, Berthold F, Arceci RJ, et al.: Clonal relationship between precursor T-lymphoblastic leukaemia/lymphoma and Langerhans-cell histiocytosis. Lancet Oncol 6 (6): 435-7, 2005.
- Rodig SJ, Payne EG, Degar BA, et al.: Aggressive Langerhans cell histiocytosis following T-ALL: clonally related neoplasms with persistent expression of constitutively active NOTCH1. Am J Hematol 83 (2): 116-21, 2008.
- Bagnasco F, Zimmermann SY, Egeler RM, et al.: Langerhans cell histiocytosis and associated malignancies: A retrospective analysis of 270 patients. Eur J Cancer 172: 138-145, 2022.
- Goyal G, Parikh R, Richman J, et al.: Spectrum of second primary malignancies and cause-specific mortality in pediatric and adult langerhans cell histiocytosis. Leuk Res 126: 107032, 2023.
Histiocitosis de células de Langerhans en adultos
La historia natural de la histiocitosis de células de Langerhans (HCL) en adultos es poco conocida. La HCL pulmonar es la excepción. En adultos el diagnóstico de esta enfermedad por lo general se retrasa muchos meses o años y se presentan problemas a largo plazo de dolor crónico y fatiga. Otras diferencias con la HCL que se presenta en la niñez, es la frecuencia de compromiso de varios sitios óseos. La HCL multisistémica de riesgo alto en adultos tal vez sea menos agresiva que la enfermedad de riesgo alto en niños. Un grupo de consenso informó sobre la evaluación y el tratamiento de los pacientes adultos con HCL. Sin embargo, el debate continúa, en especial sobre el tratamiento óptimo de primera línea.
Se realizó una revisión retrospectiva multicéntrica de 219 pacientes adultos (edad >18 años) con HCL para evaluar los desenlaces a largo plazo. La mediana de seguimiento fue de 74 meses. La tasa de supervivencia sin enfermedad a 5 años fue del 58 %, y la tasa de supervivencia general (SG) fue del 88 %. Alrededor de una tercera parte de las muertes estaban relacionadas con la HCL y se produjeron en los 5 años siguientes al diagnóstico. Se produjeron segundos cánceres en el 16,4 % de los casos (tanto hematológicos como tumores sólidos). Las muertes que se produjeron 5 años o más después del diagnóstico en su mayor parte no estuvieron relacionadas con la HCL (es decir, segundos cánceres, enfermedad pulmonar obstructiva crónica y enfermedad cardiovascular). En comparación con la población general de los Estados Unidos, los pacientes con HCL tenían una razón de mortalidad estandarizada (RME) más alta si se les diagnosticaba antes de los 55 años (RME, 5,94) o si presentaban enfermedad multisistémica (RME, 4,12).
Incidencia
En un estudio poblacional realizado en Inglaterra se encontró que la incidencia de HCL en pacientes mayores de 15 años fue de 1,05 casos por 1 millón de personas. De estas personas, el 44 % eran menores de 45 años. Una incidencia más alta de HCL en áreas económicamente desfavorecidas se relacionó con una incidencia más alta del hábito de fumar en esas áreas.
Más del 90 % de los casos de HCL pulmonar en adultos se presentan en jóvenes que fuman, a menudo más de 20 cigarrillos por día.
Cuadro clínico inicial
A menudo, los pacientes adultos exhiben signos y síntomas de HCL durante meses antes de recibir un diagnóstico definitivo y someterse a tratamiento. La HCL en adultos suele ser similar a la enfermedad en los niños y afecta los mismos órganos, aunque la incidencia en cada órgano es un poco diferente. En los adultos, predomina la enfermedad pulmonar que suele manifestarse como una enfermedad monosistémica con un vínculo estrecho con el consumo de tabaco y exhibe algunas características biológicas únicas. La mayoría de los casos de HCL pulmonar aislada en adultos son policlonales y posiblemente reactivos, mientras que son menos los casos monoclonales de HCL pulmonar.
En un registro alemán de 121 personas, se observó que el 62 % presentaba compromiso de un solo órgano y el 38 %, compromiso multisistémico. La HCL pulmonar se presentó en el 34 % de la población total del estudio. Los pulmones son el sitio más común, seguido del compromiso óseo y cutáneo. La mediana de edad en el momento del diagnóstico fue de 44 años (± 12,8 años). En estos adultos, se encontró compromiso en todos los sistemas orgánicos que se observaron en la HCL infantil, incluso el sistema endocrino y el sistema nervioso central (SNC), el hígado, el bazo, la médula ósea y el tubo digestivo. La diferencia principal fue una incidencia mucho más alta de HCL pulmonar aislada en adultos, en especial, en jóvenes fumadores. Otras diferencias fueron el compromiso más frecuente de la mucosa genital y oral.
Los signos y síntomas del cuadro clínico inicial publicados en estudios son los siguientes:
- Disnea o taquipnea.
- Polidipsia y poliuria.
- Dolor óseo.
- Edema del tejido blando cerca de las lesiones óseas.
- Exantema o nódulos en el cuero cabelludo.
- Linfadenopatía.
- Pérdida de peso.
- Fiebre.
- Hipertrofia gingival.
- Ataxia.
- Problemas de memoria.
- Hepatoesplenomegalia.
Los pacientes que presentan diabetes insípida aislada se deben observar de cerca para identificar el inicio de signos o síntomas característicos de la HCL. Al menos el 80 % de los pacientes con diabetes insípida presentaron compromiso de otros órganos, incluso huesos (68 %), piel (57 %), pulmones (39 %) y ganglios linfáticos (18 %). Sin embargo, la diabetes insípida aislada en adultos es similar a la de los pacientes pediátricos, con progresión del compromiso de la hipófisis posterior a la anterior y luego al hipotálamo y al cerebelo. Para obtener más información, consultar la sección Sistema endocrino.
Piel y cavidad oral
El compromiso cutáneo se presenta en el 37 % de los adultos con HCL multifocal. Si bien a veces hay HCL cutánea exclusiva, es menos común en los adultos que en los niños. El pronóstico de los adultos con HCL y compromiso cutáneo exclusivo es excelente; la probabilidad de supervivencia a 5 años es del 100 %. El compromiso cutáneo es similar desde el punto de vista clínico al que se observa en los niños y puede adoptar muchas formas. El compromiso inframamario y vulvar es frecuente durante el cuadro clínico inicial de mujeres adultas.
Muchos pacientes exhiben un exantema papular con áreas marrones, rojas o costrosas puntiformes de hasta un centímetro de diámetro. En el cuero cabelludo, el exantema es similar al de la seborrea. En ocasiones hay úlceras abiertas en la piel de la región inguinal, genital o perianal que no se curan después del tratamiento antibacteriano o antifúngico. En general, las lesiones son asintomáticas, pero a veces son pruriginosas o dolorosas. En la boca, la inflamación de las encías o la presencia de úlceras en las mejillas, el paladar blando o duro, las encías o la lengua son signos de HCL.
El diagnóstico de la HCL se suele confirmar mediante biopsia de la piel de lesiones cutáneas persistentes.
Huesos
La frecuencia relativa de compromiso óseo en los adultos difiere de la frecuencia en los niños. La frecuencia de compromiso mandibular es del 30 % en adultos y del 7 % en niños, y la frecuencia de compromiso craneal es del 21 % en adultos y del 40 % en niños. La frecuencia de lesiones vertebrales (13 %), pélvicas (13 %), en extremidades (17 %) y costales (6 %) en los adultos es similar a la que se encuentra en los niños.
Pulmón
La HCL pulmonar en adultos (40–50 % de los pacientes) suele ser enfermedad monosistémica. No obstante, en algunos pacientes, es posible que haya compromiso de órganos, como huesos, piel, hipotálamo o hipófisis.
La HCL pulmonar es más prevalente en fumadores que en no fumadores, y la proporción hombre-mujer es de casi 1:1, según la incidencia de consumo de cigarrillos en la población estudiada. Sin embargo, en un estudio chino sobre la HCL pulmonar, se informó que un 73 % de los pacientes eran hombres. Los pacientes con HCL pulmonar suelen presentar tos seca, disnea o dolor torácico, aunque alrededor del 20 % de los adultos con compromiso pulmonar no presentan síntomas. El dolor torácico puede indicar la presencia de un neumotórax espontáneo (10–28 % de los casos de HCL pulmonar en adultos).
Es posible diagnosticar la HCL pulmonar mediante broncoscopia en casi el 50 % de los pacientes adultos, cuando en la muestra obtenida se encuentra inmunotinción de por lo menos un 5 % de células positivas para CD1a. En la tomografía computarizada (TC) pulmonar de alta resolución se observan cambios característicos con quistes y nódulos, más prevalentes en las zonas medias y altas. Estos cambios se han caracterizado como patognomónicos para la HCL pulmonar.
En adultos, las células de la HCL en las lesiones pulmonares se ven como células dendríticas maduras que expresan índices altos de las moléculas accesorias CD80 y CD86, a diferencia de las células de Langerhans (CL) que se encuentran en otros trastornos pulmonares. Sin embargo, se han identificado variantes en la vía MAPK hasta en dos tercios de las lesiones de HCL pulmonar en adultos, lo que indica un proceso clonal en una proporción significativa de los pacientes.
En una revisión de 206 pacientes con HCL pulmonar de Francia (mediana de seguimiento, 5 años), la tasa de supervivencia a 10 años fue del 93 %. Los pacientes con insuficiencia respiratoria crónica o hipertensión pulmonar ―cada una presente en menos de un 5 % del grupo de estudio― tuvieron desenlaces mucho más precarios. El 58 % de estos pacientes murió. Los pacientes con HCL pulmonar presentaron una incidencia 17 veces más alta de carcinoma de pulmón que una cohorte de población francesa emparejada por edad y sexo.
Los factores de pronóstico favorable en la HCL pulmonar en adultos son los siguientes:
- Síntomas mínimos. Los adultos con HCL pulmonar que presentan síntomas mínimos tienen un pronóstico favorable, aunque algunos tienen un deterioro constante durante muchos años.
- Cese del consumo de tabaco o tratamiento. El 59 % de los pacientes responden de forma favorable con una remisión espontánea cuando dejan de fumar o inician algún tipo de tratamiento.Sin embargo, en un estudio, se notificó que dejar de fumar no aumentó la longevidad de los adultos con HCL pulmonar, al parecer debido a que la enfermedad exhibe una evolución muy variable. Los autores de una revisión de 206 pacientes de Francia (consultar atrás) observaron que los dos estudios citados aquí tenían menos pacientes, eran retrospectivos y en ellos no se realizaron TC de alta resolución con tanta frecuencia. Es probable que en estos estudios más antiguos se incluyeran pacientes con enfermedad más grave que en el estudio francés.
- Trasplante pulmonar. En un estudio multicéntrico, los pacientes que recibieron un trasplante de pulmón como tratamiento de la HCL pulmonar presentaron una tasa de supervivencia a 1 año del 77 % y una tasa de supervivencia a 10 años del 54 % con probabilidades de recidiva de la HCL del 20 %.
Los factores pronósticos desfavorables para la HCL pulmonar en adultos incluyen los siguientes:
- Alteración del funcionamiento pulmonar. La disminución de la proporción entre el volumen espiratorio máximo en el primer segundo y la capacidad vital forzada (VEF1/CVF), y el aumento de la proporción entre el volumen residual y la capacidad pulmonar total (VR/CPT) son variables de pronóstico adverso. Algunos pacientes tienen una función ventilatoria normal, pero una capacidad de difusión del monóxido de carbono anormal.[Nivel de evidencia C2] Alrededor del 10 % al 20 % de los pacientes presentan progresión temprana grave a insuficiencia respiratoria, hipertensión pulmonar grave y cardiopatía pulmonar. Los adultos con progresión y formación de bullas difusas, neumotórax múltiple y fibrosis tienen un pronóstico precario.; [Nivel de evidencia C2]
- Edad. Una edad mayor a 26 años es una variable de pronóstico adverso.
- Consumo de tabaco.
La mayoría de los pacientes tiene una evolución variable con enfermedad estable en algunos casos, y recaídas y progresión a disfunción respiratoria en otros casos, incluso después de muchos años. En un estudio de la historia natural de la HCL pulmonar en 58 pacientes, se encontró que el 38 % presentaban deterioro del funcionamiento pulmonar después de 2 años. Las variables de pronóstico adverso más significativas fueron el hábito de fumar y concentraciones bajas de PaO2 en el momento de la inclusión.
Se registran los siguientes resultados en las pruebas diagnósticas:
- Pruebas de funcionamiento pulmonar. El hallazgo anormal más frecuente en el funcionamiento pulmonar de los pacientes con HCL pulmonar es una reducción de la capacidad de difusión del monóxido de carbono, que se encuentra en el 70 % al 90 % de los casos.
- Tomografía computarizada. En la HCL, la TC de alta resolución revela un patrón reticulonodular con quistes y nódulos, por lo habitual en los lóbulos superiores y que no afecta el ángulo costofrénico, lo cual es característico de la HCL. La presencia de anomalías quísticas en las TC de alta resolución es un factor pronóstico precario de enfermedad progresiva.
- Biopsia. A pesar de los hallazgos característicos de la TC, la mayoría de los neumólogos están de acuerdo en que se necesita una biopsia pulmonar para confirmar el diagnóstico. En un estudio en el que se correlacionaron los hallazgos de la TC pulmonar y los resultados de la biopsia pulmonar en 27 pacientes con HCL pulmonar, se observó que los quistes de pared delgada y aspecto extraño presentaban en su interior CL y eosinófilos activos.
Hígado
En un estudio, se notificó compromiso hepático en el 27 % de los pacientes adultos con enfermedad multiorgánica. Se observó hepatomegalia (48 %) y alteración de las enzimas hepáticas (61 %). En la imágenes de TC, resonancia magnética (IRM) o ecografía a menudo se observan anomalías en el sistema biliar.
El estadio histopatológico inicial de la HCL hepática incluye la infiltración de células positivas para CD1a y fibrosis periductal con infiltrados inflamatorios y esteatosis o sin esta. El estadio tardío es la esclerosis biliar. El tratamiento con ácido ursodesoxicólico puede ser útil.
Sistema endocrino
La diabetes insípida se presenta en el 25 % de los pacientes y en ocasiones precede al diagnóstico de la HCL. Se observan anomalías en la hipófisis anterior en alrededor del 20 % de estos pacientes. A veces, los estudios por imágenes de la hipófisis son normales.
Sistema nervioso central
Las anomalías más frecuentes en el SNC son el agrandamiento de la hipófisis, su tallo o el hipotálamo. Por lo general, el compromiso encefálico se encuentra en el cerebelo, la protuberancia y los ganglios basales, con anomalías observadas en las imágenes con recuperación de la inversión atenuada de fluido (FLAIR) en T2. Algunos pacientes solo presentan cambios en las imágenes, pero otros tienen ataxia, dismetría, disartria y alteraciones conductuales y psicológicas.
Médula ósea y ganglios linfáticos
El compromiso de la médula ósea por la HCL es poco frecuente y, por lo general, se identifica por recuentos sanguíneos anormales, que también podrían ser un signo de una neoplasia maligna subyacente. La infiltración de los ganglios linfáticos por la HCL es poco frecuente como hallazgo aislado, pero se presenta en hasta un 30 % de los pacientes con HCL multisistémica.
Sistemas digestivo y cardiovascular
El compromiso digestivo es poco frecuente y, por lo general, se manifiesta con diarrea y dolor. Las anomalías en el corazón o alrededor de los grandes vasos a menudo indican una enfermedad híbrida de Erdheim-Chester (ECD) y HCL.
Enfermedad multisistémica
En una serie grande de pacientes de la Mayo Clinic, el 31 % presentó HCL multisistémica, en comparación con el 69 % de los pacientes del registro de adultos de la Histiocyte Society. Es probable que este hallazgo refleje un sesgo de derivación. En los adultos con enfermedad multisistémica, los sitios de enfermedad fueron los siguientes:
- Piel (50 %).
- Piel y mucosas (40 %).
- Hipófisis y SNC (diabetes insípida, 29,6 %).
- Hígado y bazo (hepatoesplenomegalia, 16 %).
- Glándula tiroidea (hipotiroidismo, 6,6 %).
- Ganglios linfáticos (linfadenopatía, 6 %).
Histiocitosis de células de Langerhans y neoplasias malignas relacionadas
Los adultos con HCL tienen tasas más altas de neoplasias malignas que los pacientes de la misma edad sin HCL, en una proporción de 2 a 4 según la edad del paciente. En una revisión de 132 pacientes con HCL de una sola institución, se encontraron 31 pacientes con otras neoplasias malignas antes del diagnóstico de HCL, 11 pacientes con neoplasias malignas simultáneas y 11 pacientes con otras neoplasias malignas después del diagnóstico de HCL. Los tumores sólidos representaron el 74 % de las neoplasias malignas, los linfomas el 17 % de los casos y las neoplasias malignas hematológicas el 9 % de los casos. El 71 % de los pacientes eran fumadores. Estos resultados contrastan con los de un estudio anterior basado en una revisión bibliográfica y encuestas institucionales en donde se notificó una incidencia más alta de linfomas simultáneos con el diagnóstico de HCL.
La relación entre la HCL y una neoplasia maligna se presenta con más frecuencia de lo que se esperaría por casualidad, según cuestionarios enviados a los investigadores de la Histiocyte Society y una revisión bibliográfica. En una publicación, se recopilaron casos de neoplasias malignas en pacientes con HCL entre 1991 y 2015. Se observaron 285 neoplasias malignas en 270 pacientes con HCL. Entre 154 adultos con HCL, se notificaron tumores sólidos en 61 pacientes (39,6 %), linfomas en 56 pacientes (36,4 %) y leucemias y trastornos mieloproliferativos en 37 pacientes (24,0 %). También se observaron neoplasias malignas tiroideas con cierta frecuencia. En los adultos, la HCL y las neoplasias malignas se presentaron simultáneamente en 69 pacientes (44,8 %).
En una revisión de los datos del Surveillance, Epidemiology, and End Results (SEER) Program sobre neoplasias malignas subsiguientes en 456 adultos con HCL, se encontraron 16 casos. Hubo 2 casos de linfoma no Hodgkin, 2 casos de neoplasias mielodisplásicas, 3 casos de cáncer de mama, 3 casos de cáncer de pulmón, 1 caso de cáncer colorrectal, 1 caso de cáncer de tiroides, 1 caso de cáncer de vulva, 1 caso de meningioma y 1 caso de adenocarcinoma, sin otra indicación.
En un estudio de 156 adultos con HCL se notificó la relación de la HCL con la variante BRAF V600E y la presencia de segundas neoplasias malignas primarias. Los pacientes con HCL y la variante presentaron una incidencia del 17,3 % de segundas neoplasias malignas primarias, en comparación con el 4,1 % en los pacientes sin la variante. El cociente de incidencia estandarizado (CIE) fue de 5,72 para las segundas neoplasias malignas en pacientes con HCL, en comparación con 1,7 en adultos emparejados por edad. A diferencia de los niños con HCL, no hubo correlación con la extensión de la enfermedad o la supervivencia sin progresión en adultos con variantes BRAF V600E.
Evaluación diagnóstica
La tomografía por emisión de positrones (TEP) es la modalidad más sensible para encontrar sitios afectados y es útil para el diagnóstico de la HCL. La IRM del encéfalo está indicada para pacientes con síntomas relacionados con la hipófisis y aquellos con indicios de neurodegeneración. Las IRM de la columna vertebral están indicadas para personas con dolor vertebral o neuropatía motora inferior.
adult Langerhans cell histiocytosisTratamiento de la histiocitosis de células de Langerhans en adultos
Opciones de tratamiento para la histiocitosis de células de Langerhans en adultos
La falta de ensayos clínicos limita la capacidad de emitir recomendaciones basadas en la evidencia para adultos con HCL.
Muchos investigadores han recomendado el tratamiento de acuerdo con las directrices para la HCL infantil. Sin embargo, no resulta claro si la HCL en adultos responde tan bien como la forma infantil de la enfermedad. Además, los fármacos usados en el tratamiento de niños no son tan bien tolerados cuando se administran a los adultos. Por ejemplo, la toxicidad neurológica excesiva de la vinblastina impulsó el cierre del ensayo LCH-A1. Los inhibidores de BRAF y MEK se usan cada vez más como tratamiento inicial para muchos adultos. Para obtener más información, consultar la sección Terapias dirigidas para el tratamiento de la enfermedad monosistémica y multisistémica.
Un grupo de expertos internacional propuso por consenso un algoritmo de tratamiento para adultos que se resume a continuación.
- HCL solo ósea: el tratamiento incluye curetaje, bisfosfonatos, metotrexato oral o hidroxiurea. La radioterapia se puede usar para pacientes con enfermedad progresiva y menos de 3 lesiones. Los pacientes con más de 3 lesiones pueden recibir quimioterapia (cladribina, citarabina u otros) o inhibidores de MAPK.
- HCL solo cutánea: el tratamiento incluye hidroxiurea, metotrexato oral, talidomida o lenalidomida, o terapia tópica.
- HCL pulmonar monosistémica: el tratamiento es dejar de fumar. Los pacientes con enfermedad progresiva reciben quimioterapia con fármacos similares a los que se usan para la HCL ósea.
- HCL multisistémica: el tratamiento incluye quimioterapia con fármacos similares a los que se usan para la HCL ósea.
- Compromiso de órganos vitales (médula ósea, bazo, hígado, encéfalo): los pacientes con HCL y variantes BRAF V600E reciben un inhibidor de BRAF. En los pacientes sin variantes BRAF V600E, es posible administrar un inhibidor de MEK si las pruebas son positivas para variantes de MAPK. Si no se encuentran variantes, los pacientes reciben quimioterapia según el régimen utilizado para la HCL ósea.
Tratamiento de la histiocitosis de células de Langerhans pulmonar
Es difícil determinar la eficacia de los tratamientos de la HCL pulmonar porque a veces hay resolución espontánea de la enfermedad o una enfermedad estable sin tratamiento.
Las opciones de tratamiento para los pacientes adultos con HCL pulmonar son las siguientes:
- Cese del consumo de tabaco. Dejar de fumar es obligatorio porque ese hábito tiene un efecto causal evidente en la HCL pulmonar. La mayoría de los pacientes adultos con HCL presentan progresión gradual de la enfermedad con el consumo continuado de tabaco. Es posible que la enfermedad mejore o progrese después de dejar de fumar. En un estudio de 27 pacientes con HCL pulmonar, se observó que el 52 % mejoraron después de una media de seguimiento de 14 meses. La mayoría de los pacientes mejoró al dejar de fumar y algunos mejoraron con el tratamiento con corticoesteroides. Se observó enfermedad estable en 4 pacientes (15 %) durante una media de seguimiento de 26 meses y se observó progresión de la enfermedad en 9 pacientes (33 %) durante una media de seguimiento de 22 meses.[Nivel de evidencia C3]
- Terapia con corticoesteroides. No se sabe si la terapia con corticoesteroides es eficaz para el tratamiento de la HCL pulmonar en adultos porque las series de casos notificadas no se controlaron por el cese del consumo de tabaco.
- Quimioterapia. Se notificó que algunos pacientes responden al tratamiento con cladribina o citarabina.; [Nivel de evidencia C2]
- Trasplante pulmonar. Quizás se necesite un trasplante de pulmón en los adultos que sufren extensa destrucción del tejido pulmonar debido a la HCL. En un estudio multicéntrico, se notificó una tasa de supervivencia del 54 % a los 10 años del trasplante; el 20 % de los pacientes presentó HCL recidivante que no afectó la supervivencia. Se necesita un seguimiento más prolongado de estos pacientes. En otro estudio se confirmó una tasa de supervivencia de alrededor del 50 % a los 10 años y una mejora de los cambios hemodinámicos relacionados con el tratamiento de la hipertensión arterial pulmonar, sin empeoramiento del oxígeno ni edema pulmonar.
La mejor estrategia para el seguimiento de la HCL pulmonar incluye examen físico, radiografías de tórax, pruebas de funcionamiento pulmonar y tomografías computarizadas (TC) de alta resolución.
Tratamiento de la histiocitosis de células de Langerhans ósea
Las opciones de tratamiento para los adultos con histiocitosis de células de Langerhans (HCL) ósea son las siguientes:
- Curetaje seguido de observación, con corticoesteroides intralesionales o sin estos. Al igual que en los niños, los adultos con lesiones en un solo hueso se deben someter a curetaje de la lesión seguida de observación, con corticoesteroides intralesionales o sin estos. La cirugía extensa o radical que conduce a la pérdida de funcionamiento y desfiguración está contraindicada en cualquier sitio, incluso los dientes o los huesos de la mandíbula.
- Quimioterapia sistémica. La quimioterapia sistémica produce remisión de las lesiones óseas. En el tratamiento de un número relativamente limitado de pacientes se han utilizado diversos regímenes quimioterapéuticos, como citarabina y cladribina. Para obtener más información, consultar la sección Quimioterapia y radioterapia para el tratamiento de otras enfermedades monosistémicas y multisistémicas.
- Radioterapia de dosis baja. Para los pacientes que no responden a la quimioterapia, es posible que se indique la administración de dosis bajas de radioterapia antes de cualquier cirugía radical. La radioterapia también se indica para las alteraciones neurológicas graves debido a lesión en los cuerpos vertebrales o problemas visuales por lesiones orbitarias. En 2 series y en 1 estudio, se informaron los siguientes resultados:
- En un grupo cooperativo alemán que estudió la radioterapia, se notificó una serie de 98 adultos con HCL. La mayoría de los pacientes tratados con radioterapia (60 de 98) solo tenían lesiones óseas y 24 tenían enfermedad multisistémica, incluso ósea.[Nivel de evidencia C3] De 89 pacientes evaluables, el 77 % logró una remisión completa, el 9 % presentó recidiva en el campo de radioterapia y el 15,7 % (14 de 89) presentó una progresión fuera del campo de radiación.
- En un análisis retrospectivo de 80 pacientes tratados con radioterapia sola, se notificó una tasa de remisión completa del 77 % y una tasa de remisión parcial del 12,5 %. La tasa de control a largo plazo fue del 80 % en adultos. No se notificaron efectos adversos tardíos.[Nivel de evidencia C3]
- En un estudio de una sola institución, se incluyeron 39 pacientes con HCL (intervalo de edad, 1,5–67 años; 24 pacientes de >18 años) que recibieron radioterapia para 46 lesiones. En el estudio no se notificaron recidivas locales en los 31 sitios óseos. En comparación, la tasa de ausencia de fracaso local a 3 años fue del 63 % en las 15 lesiones no óseas (intervalo de confianza 95 %, 32–83 %; P = 0,0008).
- Terapia con bisfosfonatos. En los informes de casos y series de casos se describió el uso exitoso de bisfosfonatos, tanto de pamidronato intravenoso como de zoledronato oral, para controlar el dolor óseo grave en pacientes con múltiples lesiones óseas osteolíticas de la HCL. En una revisión multiinstitucional de bisfosfonatos en niños y adultos con HCL, se encontró que la mayoría de los adultos recibieron ácido zoledrónico oral y la mayoría de los niños recibieron pamidronato.[Nivel de evidencia C3] Debido al aumento de la toxicidad de la quimioterapia en adultos, se podría usar la terapia con bisfosfonatos antes de la quimioterapia para la enfermedad ósea multifocal. Se ha notificado respuesta de otros órganos, como la piel y los tejidos blandos, a la terapia con bisfosfonatos.
- Antiinflamatorios y trofosfamida. Otro abordaje en el que se utilizaron antiinflamatorios (pioglitazona y rofecoxib) con trofosfamida en una secuencia temporal específica fue exitoso en 2 pacientes con enfermedad resistente al tratamiento con quimioterapia estándar.[Nivel de evidencia C3]
Tratamiento de la enfermedad cutánea monosistémica
Las opciones de tratamiento para los pacientes adultos con enfermedad cutánea monosistémica son las siguientes:
- Extirpación quirúrgica. Las lesiones localizadas rara vez se tratan con extirpación quirúrgica. Se debe evitar la cirugía mutiladora, incluso la hemivulvectomía, a menos que la enfermedad sea resistente a todos los tratamientos disponibles.
- Terapia tópica. Las siguientes terapias tópicas se describen en más detalle en la sección Compromiso cutáneo aislado:
- Terapia tópica o intralesional con corticoesteroides.
- Tacrólimus tópico.[Nivel de evidencia C2]
- Imiquimod tópico.[Nivel de evidencia C3]
- Psoraleno y radiación ultravioleta A de onda larga (PUVA) y UVB. Los tratamientos como PUVA/UVB pueden ser más útiles en adultos porque es posible controlar la toxicidad a largo plazo.[Nivel de evidencia C3]
- Terapia sistémica. La terapia sistémica para la HCL cutánea grave incluye metotrexato oral, hidroxiurea, talidomida oral, interferón α oral o combinaciones de interferón y talidomida.[Nivel de evidencia C3] El interferón y la talidomida también se usan para tratar la HCL cutánea crónica en adultos.[Nivel de evidencia C3] Es posible que después de suspender el tratamiento se produzcan recidivas que responden a un nuevo tratamiento.
La isotretinoína oral ha inducido remisiones en algunos adultos con HCL cutánea resistente al tratamiento.[Nivel de evidencia C3]
Quimioterapia y radioterapia para el tratamiento de otro tipo de enfermedad monosistémica y multisistémica
Evidencia (quimioterapia para el tratamiento de otro tipo de enfermedad monosistémica [no mencionada antes] y multisistémica):
- En una revisión retrospectiva de un solo centro de 58 pacientes adultos con HCL, se notificó la eficacia y toxicidad del tratamiento con vinblastina, prednisona, cladribina y citarabina.[Nivel de evidencia C3]
- Los pacientes tratados con vinblastina y prednisona presentaron el resultado más desfavorable, el 84 % de los pacientes no respondieron al cabo de 6 semanas o recayeron en el transcurso de 1 año.
- La tasa de pacientes sin respuesta o con recidiva fue del 59 % para la cladribina y del 21 % para la citarabina.
- Se presentaron efectos neurotóxicos de grado 3 o 4 en el 75 % de los pacientes tratados con vinblastina.
- Se presentó neutropenia de grado 3 o 4 en el 37 % de los pacientes tratados con cladribina y en el 20 % de los pacientes que recibieron citarabina.
- En un informe se evaluó a pacientes adultos tratados con una combinación de vindesina y prednisona o una combinación de ciclofosfamida, etopósido, vindesina y prednisona.[Nivel de evidencia C2]
- Más del 70 % de los pacientes recayeron con cualquiera de los regímenes.
- El etopósido se ha utilizado con algo de éxito en adultos con HCL monosistémica y multisistémica.
- Se notificó una toxicidad mínima con el uso prolongado de etopósido oral en adultos con HCL cutánea, mientras que los ciclos de 3 días de etopósido intravenoso (100 mg/m2/día) indujeron una remisión completa en un número pequeño de pacientes con enfermedad monosistémica y multisistémica resistente.[Nivel de evidencia C3]
- En otro estudio del mismo centro, se encontró que la azatioprina fue el fármaco más exitoso para la enfermedad localizada en adultos, con la adición de etopósido para la enfermedad resistente al tratamiento y multisistémica.[Nivel de evidencia C3]
- La cladribina es eficaz para los adultos enfermedad cutánea, ósea, ganglionar y, probablemente, enfermedad pulmonar y del SNC.[Nivel de evidencia C3];
- En un estudio multicéntrico retrospectivo, 23 pacientes que recibieron por lo menos una terapia previa se trataron con cladribina, usando varias dosis y programas de tratamiento.[Nivel de evidencia C2] La tasa de respuesta general fue del 91 % y la tasa de respuesta completa fue del 50 %. En una revisión bibliográfica, se identificaron otros 48 pacientes tratados con cladribina. En el análisis conjunto se confirmaron estos resultados.
- En un estudio se notificó sobre 38 pacientes con HCL recién diagnosticada o recidivante o resistente al tratamiento que recibieron de 1 a 9 ciclos de cladribina.[Nivel de evidencia C2] La tasa de respuesta general fue del 79 %. La tasa de respuesta completa fue del 26 % y la tasa de respuesta parcial fue del 53 %.
- Un régimen de tratamiento del linfoma usado en adultos que incluye metotrexato, doxorrubicina, ciclofosfamida, vincristina, prednisona y bleomicina (MACOP-B) se usó en 11 pacientes.
- La tasa de respuesta general fue del 100 % y la tasa de supervivencia sin progresión fue del 64 %.
- Se administró metotrexato y citarabina a 83 pacientes con HCL pulmonar (68 %), hepática (28 %), esplénica (13 %) o extrahipofisaria (4 %) recién diagnosticada.
- La tasa de respuesta objetiva fue del 88 % y un tercio de los pacientes progresó después de 3 años.
- Hubo una tasa alta de neutropenia de grados 3 a 4; casi la mitad de los pacientes presentó fiebre.
- Un tercio de los pacientes tenía trombocitopenia de grados 3 a 4.
Radioterapia. En un informe sobre la radiocirugía estereotáctica para el tratamiento de adultos con HCL hipofisaria, se observó eficacia para la reducción del tamaño de las masas.
Terapias dirigidas para el tratamiento de la enfermedad monosistémica y multisistémica
En los informes iniciales se notificó el uso de las siguientes terapias dirigidas para adultos con HCL y compromiso de sitios de riesgo bajo y alto:
- Inhibidores de la vía MAP2K/ERK. El hallazgo de que la mayoría de los pacientes con HCL tienen variantes del gen BRAF y de otros genes de la vía RAS condujo a varios informes de respuestas buenas al vemurafenib, un inhibidor de BRAF V600E, en adultos con HCL, enfermedad de Erdheim-Chester (ECD) o mezcla de ECD y HCL, así como en casos de HCL cutánea grave.[Nivel de evidencia C3]
De los 4 pacientes con HCL tratados con vemurafenib en el ensayo VE-BASKET (NCT01524978), 1 paciente presentó una respuesta completa y 3 pacientes presentaron respuestas parciales.[Nivel de evidencia C3] En 1 paciente con HCL tratado con vemurafenib mejoró la ataxia.[Nivel de evidencia C3]
En una serie, se notificaron 6 pacientes tratados con inhibidores de BRAF como tratamiento inicial. De ellos, 5 pacientes tenían enfermedad multisistémica y 1 paciente tenía HCL solo ósea. Hubo 2 respuestas completas, 3 respuestas parciales y 1 caso de enfermedad estable después de 4 a 27 meses de tratamiento.
Se llevó a cabo un ensayo clínico de prueba de concepto de cobimetinib, un inhibidor oral de MEK1 y MEK2, en 18 pacientes adultos con varios tipos de histiocitosis, incluso sarcomas histiocíticos. Los pacientes se trataron independientemente de los hallazgos genómicos. Se observaron respuestas en pacientes con variantes de ARAF, BRAF, NRAS, KRAS, MAP2K1 y MAP2K2. La tasa de respuesta general fue del 89 %, y las respuestas fueron duraderas. Al cabo de 1 año, el 94 % de los pacientes permaneció sin progresión.[Nivel de evidencia C2]
Los primeros resultados de la terapia dirigida con inhibidores son alentadores, aunque todavía hay muchas preguntas sin respuesta, en especial, con respecto a la duración óptima de la terapia y la tasa de reactivación una vez que se interrumpe la terapia. Se ha demostrado que un inhibidor de BRAF en combinación con un inhibidor de MEK es eficaz en casos de melanoma con variantes de BRAF (con toxicidad reducida). Esta combinación también puede ser eficaz en pacientes con HCL, pero no suele usarse en pacientes con enfermedades histiocíticas.[Nivel de evidencia C3] Hay varios ensayos clínicos en curso sobre inhibidores de BRAF y otros inhibidores de la vía RAS en adultos y niños con HCL.
- Otras terapias dirigidas. En un informe de casos se indica beneficio de usar infliximab, un inhibidor del factor de necrosis tumoral (FNT)-α para el tratamiento de la enfermedad neurodegenerativa en el SNC a causa de la HCL.[Nivel de evidencia C3] Sin embargo, los inhibidores del TNF infliximab y etanercept tienen una capacidad limitada para cruzar la barrera hematoencefálica. La talidomida, que también tiene actividad anti-TNF, ha sido eficaz en adultos con HCL cutánea y ósea.[Nivel de evidencia C3];
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de ensayo clínico nacional o institucional en curso:
- NCT04079179 (Cobimetinib for the Treatment of Refractory LCH): la inscripción en este estudio está abierta a niños o adultos con HCL en recaída o resistente al tratamiento o con otros trastornos histiocíticos recién diagnosticados, en recaída o resistentes al tratamiento.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
References
- Goyal G, Tazi A, Go RS, et al.: International expert consensus recommendations for the diagnosis and treatment of Langerhans cell histiocytosis in adults. Blood 139 (17): 2601-2621, 2022.
- Goyal G, Acosta-Medina AA, Abeykoon JP, et al.: Long-term outcomes among adults with Langerhans cell histiocytosis. Blood Adv 7 (21): 6568-6578, 2023.
- Liu H, Stiller CA, Crooks CJ, et al.: Incidence, prevalence and survival in patients with Langerhans cell histiocytosis: A national registry study from England, 2013-2019. Br J Haematol 199 (5): 728-738, 2022.
- Tazi A, Soler P, Hance AJ: Adult pulmonary Langerhans' cell histiocytosis. Thorax 55 (5): 405-16, 2000.
- Vassallo R, Ryu JH, Colby TV, et al.: Pulmonary Langerhans'-cell histiocytosis. N Engl J Med 342 (26): 1969-78, 2000.
- Yousem SA, Colby TV, Chen YY, et al.: Pulmonary Langerhans' cell histiocytosis: molecular analysis of clonality. Am J Surg Pathol 25 (5): 630-6, 2001.
- Roden AC, Hu X, Kip S, et al.: BRAF V600E expression in Langerhans cell histiocytosis: clinical and immunohistochemical study on 25 pulmonary and 54 extrapulmonary cases. Am J Surg Pathol 38 (4): 548-51, 2014.
- Baumgartner I, von Hochstetter A, Baumert B, et al.: Langerhans'-cell histiocytosis in adults. Med Pediatr Oncol 28 (1): 9-14, 1997.
- Kaltsas GA, Powles TB, Evanson J, et al.: Hypothalamo-pituitary abnormalities in adult patients with langerhans cell histiocytosis: clinical, endocrinological, and radiological features and response to treatment. J Clin Endocrinol Metab 85 (4): 1370-6, 2000.
- Aricò M, Girschikofsky M, Généreau T, et al.: Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer 39 (16): 2341-8, 2003.
- Götz G, Fichter J: Langerhans'-cell histiocytosis in 58 adults. Eur J Med Res 9 (11): 510-4, 2004.
- Slater JM, Swarm OJ: Eosinophilic granuloma of bone. Med Pediatr Oncol 8 (2): 151-64, 1980.
- Vassallo R, Ryu JH, Schroeder DR, et al.: Clinical outcomes of pulmonary Langerhans'-cell histiocytosis in adults. N Engl J Med 346 (7): 484-90, 2002.
- Schönfeld N, Frank W, Wenig S, et al.: Clinical and radiologic features, lung function and therapeutic results in pulmonary histiocytosis X. Respiration 60 (1): 38-44, 1993.
- Miao HL, Zhao AL, Duan MH, et al.: Clinical presentation and prognostic analysis of adult patients with Langerhans cell histiocytosis with pulmonary involvement. BMC Cancer 20 (1): 911, 2020.
- Travis WD, Borok Z, Roum JH, et al.: Pulmonary Langerhans cell granulomatosis (histiocytosis X). A clinicopathologic study of 48 cases. Am J Surg Pathol 17 (10): 971-86, 1993.
- Tazi A, Moreau J, Bergeron A, et al.: Evidence that Langerhans cells in adult pulmonary Langerhans cell histiocytosis are mature dendritic cells: importance of the cytokine microenvironment. J Immunol 163 (6): 3511-5, 1999.
- Baqir M, Vassallo R, Maldonado F, et al.: Utility of bronchoscopy in pulmonary Langerhans cell histiocytosis. J Bronchology Interv Pulmonol 20 (4): 309-12, 2013.
- Kamionek M, Ahmadi Moghaddam P, Sakhdari A, et al.: Mutually exclusive extracellular signal-regulated kinase pathway mutations are present in different stages of multi-focal pulmonary Langerhans cell histiocytosis supporting clonal nature of the disease. Histopathology 69 (3): 499-509, 2016.
- Benattia A, Bugnet E, Walter-Petrich A, et al.: Long-term outcomes of adult pulmonary Langerhans cell histiocytosis: a prospective cohort. Eur Respir J 59 (5): , 2022.
- Delobbe A, Durieu J, Duhamel A, et al.: Determinants of survival in pulmonary Langerhans' cell granulomatosis (histiocytosis X). Groupe d'Etude en Pathologie Interstitielle de la Société de Pathologie Thoracique du Nord. Eur Respir J 9 (10): 2002-6, 1996.
- Dauriat G, Mal H, Thabut G, et al.: Lung transplantation for pulmonary langerhans' cell histiocytosis: a multicenter analysis. Transplantation 81 (5): 746-50, 2006.
- Chaulagain CP: Pulmonary langerhans' cell histiocytosis. Am J Med 122 (11): e5-6, 2009.
- Lin MW, Chang YL, Lee YC, et al.: Pulmonary Langerhans cell histiocytosis. Lung 187 (4): 261-2, 2009.
- Tazi A, Hiltermann J, Vassallo R: Adult lung histiocytosis. In: Weitzman S, Egeler R M, eds.: Histiocytic Disorders of Children and Adults. Cambridge, United Kingdom: Cambridge University Press, 2005, pp 187-207.
- Tazi A, de Margerie C, Naccache JM, et al.: The natural history of adult pulmonary Langerhans cell histiocytosis: a prospective multicentre study. Orphanet J Rare Dis 10: 30, 2015.
- Crausman RS, Jennings CA, Tuder RM, et al.: Pulmonary histiocytosis X: pulmonary function and exercise pathophysiology. Am J Respir Crit Care Med 153 (1): 426-35, 1996.
- Diette GB, Scatarige JC, Haponik EF, et al.: Do high-resolution CT findings of usual interstitial pneumonitis obviate lung biopsy? Views of pulmonologists. Respiration 72 (2): 134-41, 2005 Mar-Apr.
- Soler P, Bergeron A, Kambouchner M, et al.: Is high-resolution computed tomography a reliable tool to predict the histopathological activity of pulmonary Langerhans cell histiocytosis? Am J Respir Crit Care Med 162 (1): 264-70, 2000.
- Kim HJ, Lee KS, Johkoh T, et al.: Pulmonary Langerhans cell histiocytosis in adults: high-resolution CT-pathology comparisons and evolutional changes at CT. Eur Radiol 21 (7): 1406-15, 2011.
- Abdallah M, Généreau T, Donadieu J, et al.: Langerhans' cell histiocytosis of the liver in adults. Clin Res Hepatol Gastroenterol 35 (6-7): 475-81, 2011.
- Makras P, Alexandraki KI, Chrousos GP, et al.: Endocrine manifestations in Langerhans cell histiocytosis. Trends Endocrinol Metab 18 (6): 252-7, 2007.
- Sagna Y, Courtillot C, Drabo JY, et al.: Endocrine manifestations in a cohort of 63 adulthood and childhood onset patients with Langerhans cell histiocytosis. Eur J Endocrinol 181 (3): 275-285, 2019.
- Goyal G, Hu M, Young JR, et al.: Adult Langerhans cell histiocytosis: a contemporary single-institution series of 186 patients. [Abstract] J Clin Oncol 37 (Suppl 15): A-7018, 2019. Also available online. Last accessed February 27, 2024.
- Kim HK, Park CJ, Jang S, et al.: Bone marrow involvement of Langerhans cell histiocytosis: immunohistochemical evaluation of bone marrow for CD1a, Langerin, and S100 expression. Histopathology 65 (6): 742-8, 2014.
- Singhi AD, Montgomery EA: Gastrointestinal tract langerhans cell histiocytosis: A clinicopathologic study of 12 patients. Am J Surg Pathol 35 (2): 305-10, 2011.
- Chen CY, Wu MH, Huang SF, et al.: Langerhans' cell histiocytosis presenting with a para-aortic lesion and heart failure. J Formos Med Assoc 100 (2): 127-30, 2001.
- Howarth DM, Gilchrist GS, Mullan BP, et al.: Langerhans cell histiocytosis: diagnosis, natural history, management, and outcome. Cancer 85 (10): 2278-90, 1999.
- Ma J, Laird JH, Chau KW, et al.: Langerhans cell histiocytosis in adults is associated with a high prevalence of hematologic and solid malignancies. Cancer Med 8 (1): 58-66, 2019.
- Egeler RM, Neglia JP, Puccetti DM, et al.: Association of Langerhans cell histiocytosis with malignant neoplasms. Cancer 71 (3): 865-73, 1993.
- Bagnasco F, Zimmermann SY, Egeler RM, et al.: Langerhans cell histiocytosis and associated malignancies: A retrospective analysis of 270 patients. Eur J Cancer 172: 138-145, 2022.
- Goyal G, Parikh R, Richman J, et al.: Spectrum of second primary malignancies and cause-specific mortality in pediatric and adult langerhans cell histiocytosis. Leuk Res 126: 107032, 2023.
- Acosta-Medina AA, Kemps PG, Zondag TCE, et al.: BRAF V600E is associated with higher incidence of second cancers in adults with Langerhans cell histiocytosis. Blood 142 (18): 1570-1575, 2023.
- Tazi A: Adult pulmonary Langerhans' cell histiocytosis. Eur Respir J 27 (6): 1272-85, 2006.
- Mogulkoc N, Veral A, Bishop PW, et al.: Pulmonary Langerhans' cell histiocytosis: radiologic resolution following smoking cessation. Chest 115 (5): 1452-5, 1999.
- Lorillon G, Tazi A: How I manage pulmonary Langerhans cell histiocytosis. Eur Respir Rev 26 (145): , 2017.
- Le Pavec J, Lorillon G, Jaïs X, et al.: Pulmonary Langerhans cell histiocytosis-associated pulmonary hypertension: clinical characteristics and impact of pulmonary arterial hypertension therapies. Chest 142 (5): 1150-1157, 2012.
- Abbritti M, Mazzei MA, Bargagli E, et al.: Utility of spiral CAT scan in the follow-up of patients with pulmonary Langerhans cell histiocytosis. Eur J Radiol 81 (8): 1907-12, 2012.
- Christopher Z, Binitie O, Henderson-Jackson E, et al.: Langerhans cell histiocytosis of bone in an adult: A case report. Radiol Case Rep 13 (2): 310-314, 2018.
- Cantu MA, Lupo PJ, Bilgi M, et al.: Optimal therapy for adults with Langerhans cell histiocytosis bone lesions. PLoS One 7 (8): e43257, 2012.
- Goyal G, Abeykoon JP, Hu M, et al.: Single-agent cladribine as an effective front-line therapy for adults with Langerhans cell histiocytosis. Am J Hematol 96 (5): E146-E150, 2021.
- Olschewski T, Seegenschmiedt MH: Radiotherapy of Langerhans' Cell Histiocytosis : Results and Implications of a National Patterns-of-Care Study. Strahlenther Onkol 182 (11): 629-34, 2006.
- Kriz J, Eich HT, Bruns F, et al.: Radiotherapy in langerhans cell histiocytosis - a rare indication in a rare disease. Radiat Oncol 8: 233, 2013.
- Laird J, Ma J, Chau K, et al.: Outcome After Radiation Therapy for Langerhans Cell Histiocytosis Is Dependent on Site of Involvement. Int J Radiat Oncol Biol Phys 100 (3): 670-678, 2018.
- Arzoo K, Sadeghi S, Pullarkat V: Pamidronate for bone pain from osteolytic lesions in Langerhans'-cell histiocytosis. N Engl J Med 345 (3): 225, 2001.
- Farran RP, Zaretski E, Egeler RM: Treatment of Langerhans cell histiocytosis with pamidronate. J Pediatr Hematol Oncol 23 (1): 54-6, 2001.
- Brown RE: Bisphosphonates as antialveolar macrophage therapy in pulmonary langerhans cell histiocytosis? Med Pediatr Oncol 36 (6): 641-3, 2001.
- Chellapandian D, Makras P, Kaltsas G, et al.: Bisphosphonates in Langerhans Cell Histiocytosis: An International Retrospective Case Series. Mediterr J Hematol Infect Dis 8 (1): e2016033, 2016.
- Morimoto A, Shioda Y, Imamura T, et al.: Nationwide survey of bisphosphonate therapy for children with reactivated Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer 56 (1): 110-5, 2011.
- Reichle A, Vogt T, Kunz-Schughart L, et al.: Anti-inflammatory and angiostatic therapy in chemorefractory multisystem Langerhans' cell histiocytosis of adults. Br J Haematol 128 (5): 730-2, 2005.
- Rieker J, Hengge U, Ruzicka T, et al.: [Multifocal facial eosinophilic granuloma: successful treatment with topical tacrolimus]. Hautarzt 57 (4): 324-6, 2006.
- O'Kane D, Jenkinson H, Carson J: Langerhans cell histiocytosis associated with breast carcinoma successfully treated with topical imiquimod. Clin Exp Dermatol 34 (8): e829-32, 2009.
- Taverna JA, Stefanato CM, Wax FD, et al.: Adult cutaneous Langerhans cell histiocytosis responsive to topical imiquimod. J Am Acad Dermatol 54 (5): 911-3, 2006.
- Vogel CA, Aughenbaugh W, Sharata H: Excimer laser as adjuvant therapy for adult cutaneous Langerhans cell histiocytosis. Arch Dermatol 144 (10): 1287-90, 2008.
- McClain KL, Kozinetz CA: A phase II trial using thalidomide for Langerhans cell histiocytosis. Pediatr Blood Cancer 48 (1): 44-9, 2007.
- Steen AE, Steen KH, Bauer R, et al.: Successful treatment of cutaneous Langerhans cell histiocytosis with low-dose methotrexate. Br J Dermatol 145 (1): 137-40, 2001.
- Zinn DJ, Grimes AB, Lin H, et al.: Hydroxyurea: a new old therapy for Langerhans cell histiocytosis. Blood 128 (20): 2462-2465, 2016.
- Chang SE, Koh GJ, Choi JH, et al.: Widespread skin-limited adult Langerhans cell histiocytosis: long-term follow-up with good response to interferon alpha. Clin Exp Dermatol 27 (2): 135-7, 2002.
- Tsambaos D, Georgiou S, Kapranos N, et al.: Langerhans' cell histiocytosis: complete remission after oral isotretinoin therapy. Acta Derm Venereol 75 (1): 62-4, 1995.
- Duan MH, Han X, Li J, et al.: Comparison of vindesine and prednisone and cyclophosphamide, etoposide, vindesine, and prednisone as first-line treatment for adult Langerhans cell histiocytosis: A single-center retrospective study. Leuk Res 42: 43-6, 2016.
- Tsele E, Thomas DM, Chu AC: Treatment of adult Langerhans cell histiocytosis with etoposide. J Am Acad Dermatol 27 (1): 61-4, 1992.
- Chu T: Langerhans cell histiocytosis. Australas J Dermatol 42 (4): 237-42, 2001.
- Saven A, Foon KA, Piro LD: 2-Chlorodeoxyadenosine-induced complete remissions in Langerhans-cell histiocytosis. Ann Intern Med 121 (6): 430-2, 1994.
- Pardanani A, Phyliky RL, Li CY, et al.: 2-Chlorodeoxyadenosine therapy for disseminated Langerhans cell histiocytosis. Mayo Clin Proc 78 (3): 301-6, 2003.
- Néel A, Artifoni M, Fontenoy AM, et al.: Long-term efficacy and safety of 2CdA (cladribine) in extra-pulmonary adult-onset Langerhans cell histiocytosis: analysis of 23 cases from the French Histiocytosis Group and systematic literature review. Br J Haematol 189 (5): 869-878, 2020.
- Derenzini E, Stefoni V, Pellegrini C, et al.: High efficacy of the MACOP-B regimen in the treatment of adult Langerhans cell histiocytosis, a 20 year experience. BMC Cancer 15: 879, 2015.
- Cao XX, Li J, Zhao AL, et al.: Methotrexate and cytarabine for adult patients with newly diagnosed Langerhans cell histiocytosis: A single arm, single center, prospective phase 2 study. Am J Hematol 95 (9): E235-E238, 2020.
- Hong WC, Murovic JA, Gibbs I, et al.: Pituitary stalk Langerhans cell histiocytosis treated with CyberKnife radiosurgery. Clin Neurol Neurosurg 115 (5): 573-7, 2013.
- Haroche J, Cohen-Aubart F, Emile JF, et al.: Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 121 (9): 1495-500, 2013.
- Charles J, Beani JC, Fiandrino G, et al.: Major response to vemurafenib in patient with severe cutaneous Langerhans cell histiocytosis harboring BRAF V600E mutation. J Am Acad Dermatol 71 (3): e97-9, 2014.
- Diamond EL, Durham BH, Ulaner GA, et al.: Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 567 (7749): 521-524, 2019.
- Diamond EL, Subbiah V, Lockhart AC, et al.: Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncol 4 (3): 384-388, 2018.
- Hyman DM, Puzanov I, Subbiah V, et al.: Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med 373 (8): 726-36, 2015.
- Hazim AZ, Ruan GJ, Ravindran A, et al.: Efficacy of BRAF-Inhibitor Therapy in BRAFV600E -Mutated Adult Langerhans Cell Histiocytosis. Oncologist 25 (12): 1001-1004, 2020.
- Chohan G, Barnett Y, Gibson J, et al.: Langerhans cell histiocytosis with refractory central nervous system involvement responsive to infliximab. J Neurol Neurosurg Psychiatry 83 (5): 573-5, 2012.
- Sander CS, Kaatz M, Elsner P: Successful treatment of cutaneous langerhans cell histiocytosis with thalidomide. Dermatology 208 (2): 149-52, 2004.
- Crickx E, Bouaziz JD, Lorillon G, et al.: Clinical Spectrum, Quality of Life, BRAF Mutation Status and Treatment of Skin Involvement in Adult Langerhans Cell Histiocytosis. Acta Derm Venereol 97 (7): 838-842, 2017.
Actualizaciones más recientes a este resumen (07/05/2024)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Características histopatológicas, inmunológicas y citogenéticas de la histiocitosis de células de Langerhans
Se añadió texto sobre los resultados de un estudio en el que se usó un modelo murino con la variante BRAF V600E bajo el control de los promotores de los genes Scl o Map17 y que añadió conocimientos adicionales sobre las características biológicas de la HCL neurodegenerativa (se citó a Wilk et al. como referencia 31).
Histiocitosis de células de Langerhans infantil
Se añadió texto para indicar que, aunque se han notificado neoplasias malignas, como el carcinoma de células escamosas en adultos tratados con inhibidores de MAPK, no se han notificado dichas neoplasias en pacientes pediátricos. Al igual que los adultos, los niños desarrollan erupciones acneiformes, fotosensibilidad, diarrea y, a veces, mialgias.
Se añadió texto para indicar que, en informes de casos y series de casos, se ha documentado la eficacia de los inhibidores de MAPK para el tratamiento de la HCL hepática progresiva (se citó a Lee et al. como referencia 159).
Se añadió NCT04079179 como ensayo en el que la inscripción en este estudio está abierta a niños o adultos con HCL en recaída o resistente al tratamiento o con otros trastornos histiocíticos recién diagnosticados, en recaída o resistentes al tratamiento.
Se revisó el texto para indicar que la evaluación de la respuesta es más fácil cuando hay un área específica, que se puede controlar por evaluación clínica o con ecografía, tomografías computarizadas, tomografías por emisión de positrones o imágenes por resonancia magnética, como la piel, la hepatomegalia o la esplenomegalia y otras lesiones expansivas o líticas óseas.
Se revisó el texto para indicar que los pacientes con compromiso multisistémico tienen una incidencia de complicaciones a largo plazo de cerca del 70 %. Sin embargo, no se ha informado del alcance de las secuelas a largo plazo en pacientes que reciben tratamiento durante un año.
Histiocitosis de células de Langerhans en adultos
Se añadió texto para indicar que el debate sobre el tratamiento continúa, en especial sobre el tratamiento óptimo de primera línea para adultos con HCL.
Se añadió texto sobre los resultados una revisión retrospectiva multicéntrica de 219 pacientes adultos con HCL para evaluar los desenlaces a largo plazo (se citó a Goyal et al. como referencia 2).
Se añadió Opciones de tratamiento en evaluación clínica como subsección nueva.
El Consejo editorial del PDQ sobre el tratamiento pediátrico es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este resumen del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento de la histiocitosis de células de Langerhans en niños y adultos. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El consejo editorial del PDQ sobre el tratamiento pediátrico, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
- Si el artículo se debe analizar en una reunión del consejo.
- Si conviene añadir texto acerca del artículo.
- Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Tratamiento de la histiocitosis de células de Langerhans son:
- Louis S. Constine, MD (James P. Wilmot Cancer Center at University of Rochester Medical Center)
- Thomas G. Gross, MD, PhD (National Cancer Institute)
- Michael Jeng, MD (Stanford Medicine Children's Health)
- Kenneth L. McClain, MD, PhD (Texas Children's Cancer Center and Hematology Service at Texas Children's Hospital)
- Carlos Rodriguez-Galindo, MD (St. Jude Children's Research Hospital)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El consejo editorial del PDQ sobre el tratamiento pediátrico emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como “En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]”.
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
PDQ® sobre el tratamiento pediátrico. PDQ Tratamiento de la histiocitosis de células de Langerhans. Bethesda, MD: National Cancer Institute. Actualización:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como “estándar” o “en evaluación clínica”. Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Comuníquese con el Instituto Nacional del Cáncer
Para obtener más información sobre las opciones para comunicarse con el NCI, incluso la dirección de correo electrónico, el número telefónico o el chat, consultar la página del Servicio de Información de Cáncer del Instituto Nacional del Cáncer.