Instituto Nacional del Cáncer
encontrar mi
Fecha de publicación: Mar 14, 2024
Información sobre el cáncer de hígado en los niños y las opciones de tratamiento: cirugía, quimioterapia, radiación, quimioembolización y radioembolización. Resumen para profesionales de la salud.
Tratamiento del cáncer de hígado infantil
Información general sobre el cáncer de hígado infantil
El cáncer de hígado (cáncer hepático) es una neoplasia maligna infrecuente en niños y adolescentes que se divide en los siguientes dos subgrupos histológicos principales:
- Hepatoblastoma.
- Subtipo histológico fetal bien diferenciado (fetal puro).
- Subtipo histológico epitelial y fetal mixto.
- Hepatoblastoma de subtipo histológico indiferenciado de células pequeñas y tumores rabdoides de hígado.
- Hepatoblastoma indiferenciado de células pequeñas (positivo para SMARCB1).
- Tumor rabdoide de hígado (negativo para SMARCB1).
- Carcinoma hepatocelular.
Otros tipos histológicos menos comunes son los siguientes:
- Sarcoma embrionario indiferenciado de hígado.
- Coriocarcinoma de hígado infantil (de lactantes).
- Tumores vasculares hepáticos.
- Rabdomiosarcoma biliar. Para obtener más información, consultar Tratamiento del rabdomiosarcoma infantil.
Clasificación celular del cáncer de hígado infantil
Los tumores de hígado son infrecuentes en la niñez. El diagnóstico a veces presenta un reto; en parte, debido a la falta de consenso respecto a un sistema de clasificación. Una revisión histopatológica central sistemática de estos tumores realizada como parte de protocolos terapéuticos colaborativos pediátricos, permitió identificar subtipos histológicos asociados con manifestaciones clínicas específicas. Como resultado, las características histopatológicas se incorporaron en los protocolos del Children’s Oncology Group (COG) y, en los Estados Unidos, son un parámetro de estratificación del riesgo para el tratamiento del paciente.
El COG Liver Tumor Committee patrocinó el International Pathology Symposium en 2011 donde se analizaron las características histopatológicas y la clasificación de los tumores de hígado en pediatría (en especial, el hepatoblastoma) y se formuló una clasificación llamada International Pediatric Liver Tumors Consensus Classification que es de uso obligado para los proyectos colaborativos internacionales. Los resultados de esta clasificación internacional de tumores de hígado en pediatría ya se publicaron. Esta clasificación estandarizada y clínicamente significativa permitirá la integración de nuevos parámetros biológicos y las características genéticas tumorales dentro de un lenguaje patológico común para ayudar a mejorar el tratamiento y los desenlaces de los pacientes en el futuro.
Para obtener información sobre las características histológicas de cada subtipo de cáncer de hígado infantil, consultar las siguientes secciones:
- Hepatoblastoma.
- Carcinoma hepatocelular.
- Sarcoma embrionario indiferenciado de hígado.
- Coriocarcinoma de hígado infantil (de lactantes).
- Tumores vasculares hepáticos.
References
- López-Terrada D, Alaggio R, de Dávila MT, et al.: Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol 27 (3): 472-91, 2014.
Estratificación tumoral por imágenes y estadificación quirúrgica de Evans para el cáncer de hígado infantil
Tradicionalmente, los 4 grupos principales que estudian este tema (el International Childhood Liver Tumors Strategy Group [antes conocido como Société Internationale d’Oncologie Pédiatrique–Epithelial Liver Tumor Study Group (SIOPEL)], el Children's Oncology Group [COG], la Gesellschaft für Pädiatrische Onkologie und Hämatologie [Society for Paediatric Oncology and Haematology] y el Japanese Study Group for Pediatric Liver Tumors) usaban diferentes categorías de estratificación del riesgo, lo que dificultaba la comparación transcontinental de los resultados. Ahora, todos los grupos están usando el sistema de agrupación PRE-Treatment EXTent of tumor (PRETEXT) como parte de la estratificación del riesgo.
Estratificación del tumor por imágenes
El objetivo principal del tratamiento de los pacientes con cáncer de hígado es la extirpación quirúrgica del tumor primario. Por lo tanto, la agrupación por riesgo depende en gran medida de factores determinados con imágenes que están relacionados con una resección quirúrgica inocua del tumor, así como de la agrupación PRETEXT. Estos hallazgos de las imágenes incluyen la sección o secciones del hígado afectadas por el tumor y otros hallazgos, denominados factores de anotación, que inciden en la toma de decisiones quirúrgicas y en el pronóstico.
La estratificación del riesgo de los niños con hepatoblastoma depende del uso de imágenes transversales de calidad alta. Para obtener las imágenes, se usan la tomografía trifásica computarizada (sin contraste, arterial y venosa) o las imágenes por resonancia magnética (IRM) con contraste. La IRM con gadoxetato disódico, un fármaco a base de gadolinio que los hepatocitos absorben y excretan preferentemente, se usa cada vez más y puede mejorar la detección de la enfermedad multifocal.
Definiciones de los grupos PRETEXT y POSTTEXT
Los sistemas de agrupación por imágenes que se usan para definir radiológicamente la extensión del compromiso tumoral en el hígado son los siguientes:
- PRETEXT (PRE-Treatment EXTent of disease). La extensión del compromiso hepático se define antes del tratamiento.
- POSTTEXT (POST-Treatment EXTent of disease). La extensión del compromiso hepático se define en respuesta al tratamiento.
PRETEXT
PRETEXT se usa en los principales grupos de ensayos multicéntricos como componente central de los esquemas de estratificación del riesgo que definen el tratamiento del hepatoblastoma. PRETEXT se basa en la estructura anatómica de Couinaud de 8 segmentos del hígado evaluados en imágenes transversales.
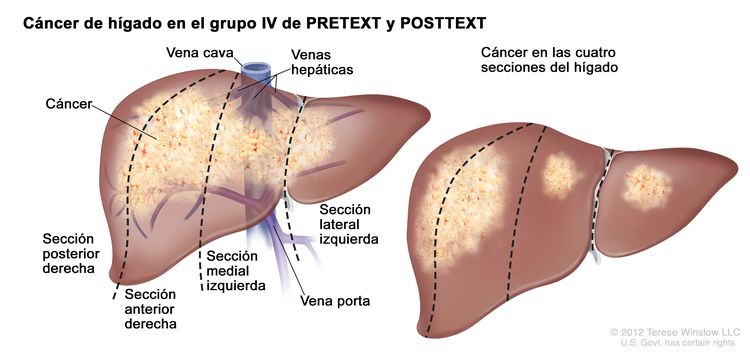
El sistema PRETEXT divide el hígado en 4 partes llamadas secciones. El lóbulo izquierdo del hígado se compone de una sección lateral (segmentos de Couinaud I, II y III) y una sección medial (segmento IV), mientras que el lóbulo derecho se compone de una sección anterior (segmentos V y VIII) y una sección posterior (segmentos VI y VII); (consultar la Figura 1). Los grupos PRETEXT fueron ideados por SIOPEL para su primer ensayo, SIOPEL-1, Y revisado para el ensayo SIOPEL-3 en 2007.
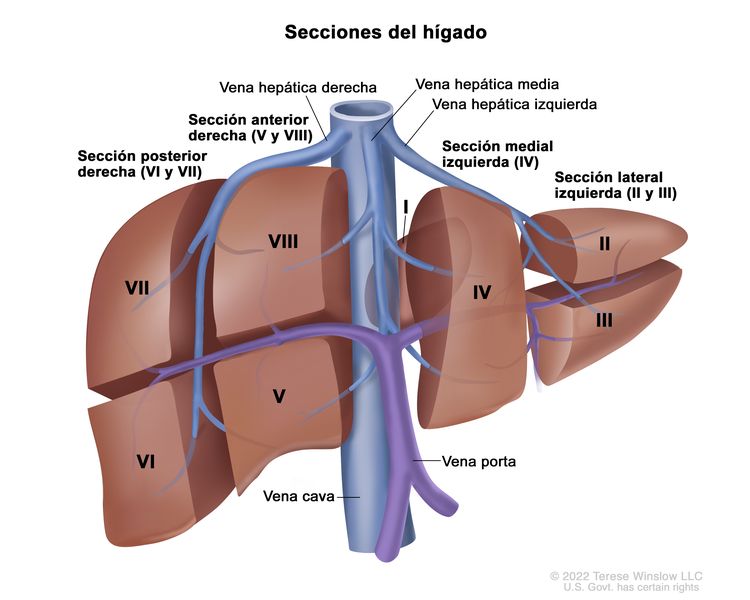 Figura 1. El hígado está dividido en 4 secciones: la sección posterior derecha, la sección anterior derecha, la sección medial izquierda y la sección lateral izquierda. Además, cada sección del hígado se divide en segmentos. Los segmentos VI y VII forman la sección posterior derecha, los segmentos V y VIII forman la sección anterior derecha, el segmento IV forma la sección medial izquierda, y los segmentos II y III forman la sección lateral izquierda. El segmento I tiene una ubicación más profunda en el lado izquierdo del hígado, delante de la vena cava inferior y por detrás de las venas hepáticas derecha, media e izquierda.
Figura 1. El hígado está dividido en 4 secciones: la sección posterior derecha, la sección anterior derecha, la sección medial izquierda y la sección lateral izquierda. Además, cada sección del hígado se divide en segmentos. Los segmentos VI y VII forman la sección posterior derecha, los segmentos V y VIII forman la sección anterior derecha, el segmento IV forma la sección medial izquierda, y los segmentos II y III forman la sección lateral izquierda. El segmento I tiene una ubicación más profunda en el lado izquierdo del hígado, delante de la vena cava inferior y por detrás de las venas hepáticas derecha, media e izquierda.
La asignación al grupo PRETEXT I, II, III, o IV se determina por el número de secciones del hígado sin compromiso. La clasificación PRETEXT se complementa con los factores de anotación. Los factores de anotación incluyen hallazgos que son importantes para el tratamiento quirúrgico e indicios de extensión tumoral fuera del parénquima de las secciones principales del hígado, incluso la enfermedad metastásica. Para obtener una descripción detallada de los grupos PRETEXT, consultar el Cuadro 1. Para obtener descripciones de los factores de anotación, consultar el Cuadro 2.
Los factores de anotación identifican el grado de compromiso tumoral de los vasos principales y su efecto en el flujo venoso de entrada y salida, que es información esencial para el cirujano y puede afectar los desenlaces quirúrgicos. El COG y los principales centros de cirugía hepática en los Estados Unidos usaban diferentes definiciones del compromiso vascular macroscópico en comparación con las definiciones de SIOPEL que se usaban en Europa. Estas diferencias se han resuelto y las nuevas definiciones se están utilizando en un ensayo internacional que comenzó en 2018.
Si bien PRETEXT se usa para predecir la resecabilidad del tumor, tiene limitaciones. Es difícil diferenciar la invasión real más allá del borde anatómico de una sección hepática determinada de la compresión y el desplazamiento causados por el tumor; sobre todo, en el momento del diagnóstico. Además, en ocasiones es difícil distinguir entre la coartación vascular y el compromiso vascular, en especial si no se usa la imagen adecuada. La asignación al grupo PRETEXT tiene un grado moderado de variabilidad entre observadores. En un informe publicado en 2005 con datos del estudio SIOPEL-1, el grupo preoperatorio PRETEXT se alineó con los hallazgos patológicos posoperatorios solo el 51 % de las veces, con sobreestadificación en el 37 % de los pacientes y subestadificación en el 12 % de los pacientes.
Debido a que es difícil distinguir la asignación al grupo PRETEXT, la revisión central de las imágenes es de suma importancia y se suele hacer en todos los ensayos clínicos importantes. Para los pacientes que no participan en ensayos clínicos, se debe considerar la revisión radiológica experta en los casos cuestionables cuya asignación al grupo PRETEXT afecta la elección del tratamiento.
| Grupos PRETEXT y POSTTEXT | Definición | Imagen | |
|---|---|---|---|
| aAdaptación de Roebuck et al. | |||
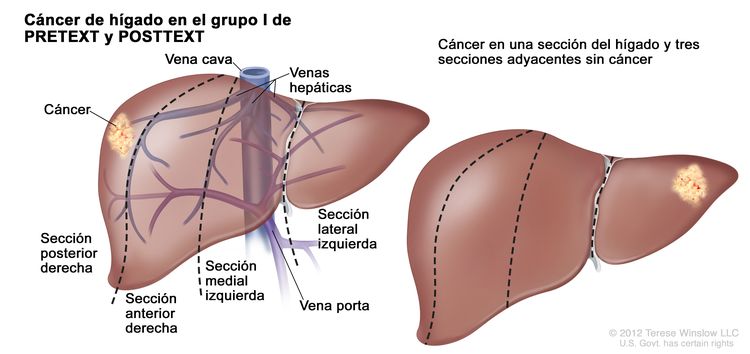
| I | Compromiso de 1 sola sección; 3 secciones adyacentes sin tumor. |
| |
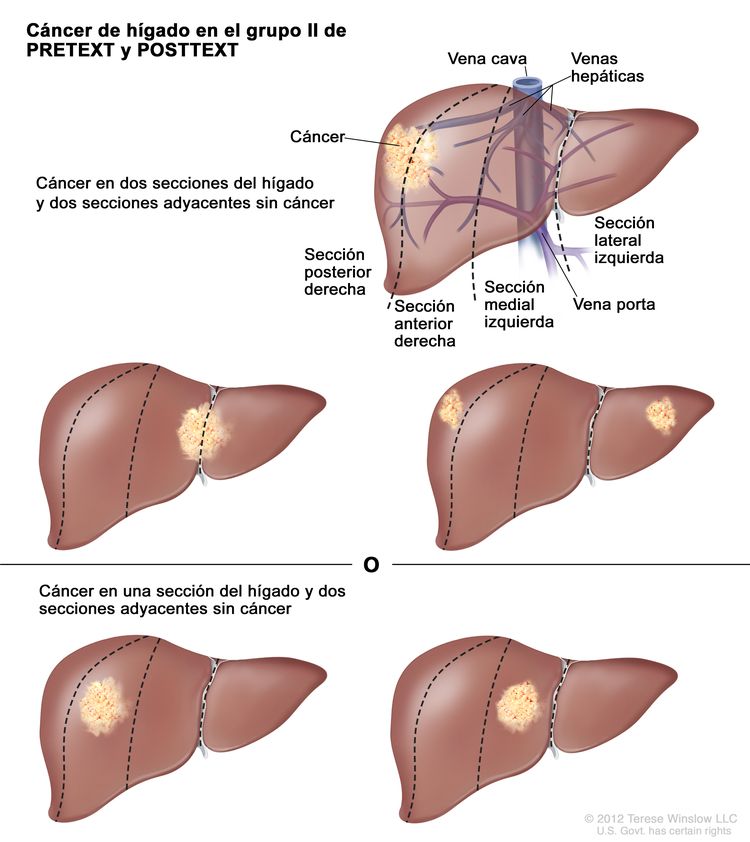
| II | Compromiso de 1 o 2 secciones; 2 secciones adyacentes sin tumor. |
| |
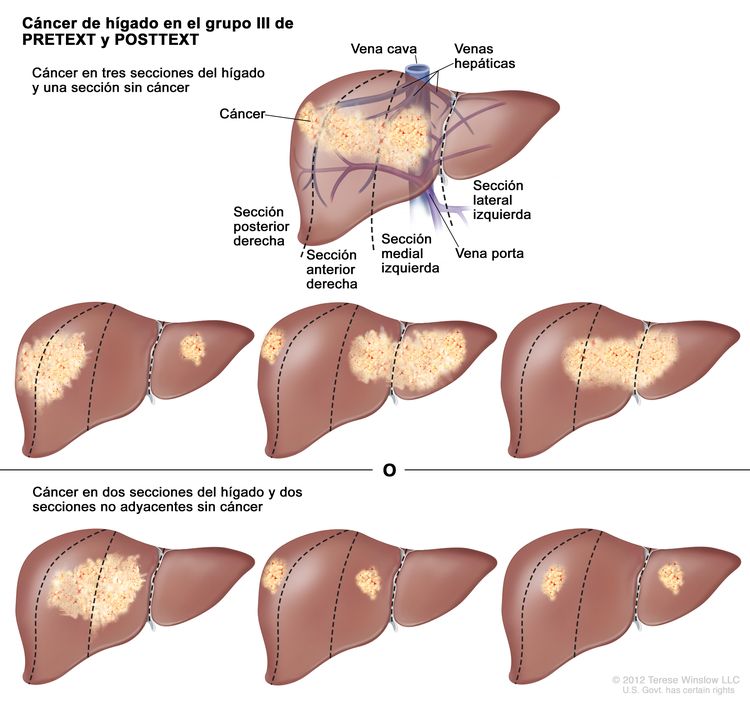
| III | Compromiso de 2 o 3 secciones; 1 sección adyacente sin tumor. |
| |
| IV | Compromiso de 4 secciones. |
| |
| Factores de anotación | Definición | |||
|---|---|---|---|---|
| TC = tomografía computarizada; IRM = imágenes por resonancia magnética; UH = unidad de Hounsfield. | ||||
| aAdaptación de Roebuck et al. | ||||
| bSe publicaron detalles adicionales que describen los factores de anotación. | ||||
| Vb | Compromiso venoso: compromiso vascular de la vena cava retrohepática o compromiso de las 3 venas hepáticas principales (derecha, media e izquierda). | |||
| V0 | Tumor a menos de 1 cm del vaso. | |||
| V1 | Tumor adyacente al vaso. | |||
| V2 | Tumor que comprime o distorsiona el vaso. | |||
| V3 | Crecimiento tumoral infiltrante, atrapamiento vascular o trombo. | |||
| Pb | Compromiso portal: compromiso vascular de la vena porta principal o de ambas venas portas, derecha e izquierda. | |||
| P0 | Tumor a menos de 1 cm del vaso. | |||
| P1 | Tumor adyacente a la vena porta principal, las venas portas derecha e izquierda o la bifurcación de la vena porta. | |||
| P2 | Tumor que comprime la vena porta principal, las venas portas derecha e izquierda o la bifurcación de la vena porta. | |||
| P3 | Crecimiento tumoral infiltrante, atrapamiento vascular (>50 % o >180 grados) o trombo intravascular en la vena porta principal, las venas portas derecha e izquierda o la bifurcación de la vena porta. | |||
| Eb | Diseminación extrahepática de la enfermedad. Se cumple cualquiera de los siguientes criterios: | |||
| E1 | Tumor que cruza los límites o los planos tisulares. | |||
| E2 | Tumor rodeado de tejido normal en más de 180 grados. | |||
| E3 | Presencia de nódulos peritoneales (que no son ganglios linfáticos); al menos 1 nódulo ≥10 mm o 2 nódulos ≥5 mm. | |||
| Mb | Metástasis a distancia. Se cumple cualquiera de los siguientes criterios: | |||
| M1 | 1 nódulo pulmonar no calcificado de diámetro ≥5 mm. | |||
| M2 | 2 o más nódulos no calcificados, cada uno de diámetro ≥3 mm. | |||
| M3 | Enfermedad metastásica confirmada mediante estudio patológico. | |||
| C | Tumor que compromete el lóbulo caudado. | |||
| F | Multifocalidad: 2 o más tumores hepáticos aislados con tejido hepático intermedio normal. | |||
| Nb | Metástasis ganglionares. Se cumple cualquiera de los siguientes criterios: | |||
| N1 | Ganglio linfático con un diámetro de eje corto >1 cm. | |||
| N2 | Ganglio linfático portocavo con un diámetro de eje corto >1,5 cm. | |||
| N3 | Ganglio linfático esférico con pérdida del hilio graso. | |||
| Rb | Ruptura del tumor. Líquido libre en el abdomen o la pelvis con uno o más de los siguientes signos de hemorragia: | |||
| R1 | Complejidad interna o tabicaciones que separan el líquido. | |||
| R2 | Se observa líquido de alta densidad en la TC (>25 UH). | |||
| R3 | Se observan imágenes características de sangre o de productos de degradación de la sangre en la IRM. | |||
| R4 | Se observa líquido heterogéneo con partículas ecogénicas en la ecografía. | |||
| R5 | Cápsula tumoral con anomalía visible o células tumorales en el líquido peritoneal o ruptura diagnosticada mediante estudio patológico en pacientes que se sometieron a una resección inicial. | |||
POSTTEXT
El grupo POSTTEXT se determina después de que los pacientes reciben quimioterapia. La respuesta más alta a la quimioterapia, medida como disminución en el tamaño del tumor y la concentración de alfafetoproteína (AFP), se produce después de los 2 primeros ciclos de quimioterapia. Además, en un estudio en el que se evaluó la resecabilidad quirúrgica después de 2 versus 4 ciclos de quimioterapia, se observó que muchos tumores se pueden ser resecar después de 2 ciclos.
Estadificación quirúrgica de Evans para el cáncer de hígado infantil (histórica)
El sistema de estadificación de Evans y del COG, basado en los hallazgos operatorios y la resecabilidad quirúrgica, se utilizó durante muchos años en los Estados Unidos para agrupar y determinar el tratamiento de los niños con cáncer de hígado (consultar el Cuadro 3). En la actualidad, se utilizan otros sistemas de estratificación del riesgo para clasificar a los pacientes y determinar la estrategia de tratamiento. Para obtener más información, consultar el Cuadro 5.
| Estadio quirúrgico de Evans | Definición |
|---|---|
| Estadio I | El tumor se resecó por completo. |
| Estadio II | Queda tumor residual microscópico después de la resección. |
| Estadio III | No hay metástasis a distancia y se presenta al menos una de las siguientes situaciones: 1) El tumor es irresecable, o se extirpó pero quedan residuos tumorales macroscópicos; 2) hay compromiso de ganglios linfáticos extrahepáticos. |
| Estadio IV | Hay metástasis a distancia, con independencia del grado de compromiso hepático. |
References
- Meyers AB, Towbin AJ, Geller JI, et al.: Hepatoblastoma imaging with gadoxetate disodium-enhanced MRI--typical, atypical, pre- and post-treatment evaluation. Pediatr Radiol 42 (7): 859-66, 2012.
- Brown J, Perilongo G, Shafford E, et al.: Pretreatment prognostic factors for children with hepatoblastoma-- results from the International Society of Paediatric Oncology (SIOP) study SIOPEL 1. Eur J Cancer 36 (11): 1418-25, 2000.
- Roebuck DJ, Aronson D, Clapuyt P, et al.: 2005 PRETEXT: a revised staging system for primary malignant liver tumours of childhood developed by the SIOPEL group. Pediatr Radiol 37 (2): 123-32; quiz 249-50, 2007.
- Towbin AJ, Meyers RL, Woodley H, et al.: 2017 PRETEXT: radiologic staging system for primary hepatic malignancies of childhood revised for the Paediatric Hepatic International Tumour Trial (PHITT). Pediatr Radiol 48 (4): 536-554, 2018.
- Aronson DC, Schnater JM, Staalman CR, et al.: Predictive value of the pretreatment extent of disease system in hepatoblastoma: results from the International Society of Pediatric Oncology Liver Tumor Study Group SIOPEL-1 study. J Clin Oncol 23 (6): 1245-52, 2005.
- Lovvorn HN, Ayers D, Zhao Z, et al.: Defining hepatoblastoma responsiveness to induction therapy as measured by tumor volume and serum alpha-fetoprotein kinetics. J Pediatr Surg 45 (1): 121-8; discussion 129, 2010.
- Venkatramani R, Stein JE, Sapra A, et al.: Effect of neoadjuvant chemotherapy on resectability of stage III and IV hepatoblastoma. Br J Surg 102 (1): 108-13, 2015.
- Ortega JA, Krailo MD, Haas JE, et al.: Effective treatment of unresectable or metastatic hepatoblastoma with cisplatin and continuous infusion doxorubicin chemotherapy: a report from the Childrens Cancer Study Group. J Clin Oncol 9 (12): 2167-76, 1991.
- Douglass EC, Reynolds M, Finegold M, et al.: Cisplatin, vincristine, and fluorouracil therapy for hepatoblastoma: a Pediatric Oncology Group study. J Clin Oncol 11 (1): 96-9, 1993.
- Ortega JA, Douglass EC, Feusner JH, et al.: Randomized comparison of cisplatin/vincristine/fluorouracil and cisplatin/continuous infusion doxorubicin for treatment of pediatric hepatoblastoma: A report from the Children's Cancer Group and the Pediatric Oncology Group. J Clin Oncol 18 (14): 2665-75, 2000.
Aspectos generales de las opciones de tratamiento del cáncer de hígado infantil
Muchas de las mejoras en la supervivencia del cáncer infantil se lograron con el uso de tratamientos nuevos en los que se intentó superar el mejor tratamiento disponible ya aceptado. Los ensayos clínicos de pediatría se diseñan a fin de comparar un tratamiento que parece mejor con el tratamiento estándar actual. Es posible realizar esta comparación en un ensayo aleatorizado con dos grupos de tratamiento o mediante la evaluación de un solo tratamiento nuevo que se compara con los resultados obtenidos antes con el tratamiento estándar.
Debido a que el cáncer en los niños es relativamente poco frecuente, todos los niños con cáncer de hígado se consideran aptos para participar en un ensayo clínico siempre que esté disponible. Para determinar y poner en práctica un tratamiento óptimo, es necesario que la planificación esté a cargo de un equipo multidisciplinario de especialistas en cáncer con experiencia en el tratamiento de tumores infantiles.
Cirugía
Históricamente, la resección quirúrgica completa del tumor primario ha sido esencial para la curación de los tumores malignos de hígado en los niños.; [Nivel de evidencia C1] Este abordaje sigue siendo el objetivo de los procedimientos quirúrgicos definitivos, que a menudo se combinan con quimioterapia. El cirujano realiza una resección hepática muy sofisticada en niños y adolescentes con tumores hepáticos primarios después de confirmar el diagnóstico mediante los estudios patológicos de las secciones congeladas intraoperatorias. La resección quirúrgica completa es importante para todos los tumores de hígado, en especial para los carcinomas hepatocelulares debido a que no se dispone de quimioterapia curativa. En los pacientes con hepatoblastoma avanzado, las complicaciones posoperatorias se relacionan con un empeoramiento de la supervivencia general (SG).
Las tres opciones quirúrgicas para tratar el cáncer primario de hígado en pediatría son las siguientes:
- Resección quirúrgica inicial (sola o con quimioterapia adyuvante).
- Resección quirúrgica diferida (con quimioterapia neoadyuvante).
- Trasplante ortotópico de hígado (donante cadavérico o vivo) (casi siempre precedido de quimioterapia).
La decisión sobre qué método quirúrgico usar (por ejemplo, hepatectomía parcial, resección amplia o trasplante) depende de muchos factores como los siguientes:
- Grupo de PRE-Treatment EXTent of disease (PRETEXT) y POST-Treatment EXTent of disease (POSTTEXT).
- Tamaño del tumor primario.
- Presencia de enfermedad hepática multifocal.
- Compromiso vascular macroscópico.
- Concentraciones de alfafetoproteína (AFP).
- Si es posible que la quimioterapia preoperatoria convierta un tumor irresecable en uno resecable.
- Si la enfermedad hepática cumple con los criterios quirúrgicos e histopatológicos para un trasplante ortotópico de hígado.
Determinar el momento oportuno del abordaje quirúrgico es de suma importancia. Por esta razón, los cirujanos con experiencia en resecciones quirúrgicas hepáticas y trasplantes en el ámbito pediátrico participan desde el inicio en la toma de decisiones para determinar el momento oportuno y el alcance de la resección.
La participación temprana, de manera preferente en el momento del diagnóstico, de un cirujano pediatra experto en cirugía hepática es importante, en especial, en pacientes de los grupos PRETEXT III o IV o quienes tienen compromiso de los vasos hepáticos mayores (factores de anotación de compromiso V [venoso] o P [portal]). Aunque inicialmente se pensó que el compromiso vascular era una contraindicación para la resección, los cirujanos hepáticos con bastante experiencia son capaces de resecar el tumor de manera exitosa y pueden evitar un trasplante.; [Nivel de evidencia C1] Los pacientes con compromiso vascular y tumores que el experto en cirugía pediátrica consideró no resecables se deben derivar a un centro de trasplante para su evaluación a fin de evitar retrasos innecesarios en la evaluación y la lista de pacientes para trasplante.
Es posible que una ecografía intraoperatoria permita delinear aún más la extensión y localización del tumor, y de esta manera incidir en el abordaje intraoperatorio. Además se ha utilizado una infusión preoperatoria de verde de indocianina, una sustancia fluoroactiva que se concentra en el hígado y se acumula en los tumores hepáticos anormales, con el fin de proporcionar una orientación visual intraoperatoria de la ubicación del tumor y para evaluar la proximidad a los márgenes quirúrgicos.
Si se determina que el tumor es irresecable, se deben considerar las medidas para reducir el tamaño del tumor de manera que sea posible una resección quirúrgica completa. Estas medidas incluyen la quimioterapia intravenosa preoperatoria, la quimioterapia transarterial o la terapia radiactiva transarterial. Estos esfuerzos se deben coordinar de manera minuciosa con el equipo de cirugía para facilitar la planificación de la resección. El uso prolongado de quimioterapia en ocasiones conduce a retrasos innecesarios y, en casos poco frecuentes, a la progresión tumoral. Si un equipo quirúrgico experimentado logra extirpar todo el tumor, es posible que se necesite menos quimioterapia posoperatoria. Se debe evitar la resección incompleta porque los pacientes que se someten a trasplantes de rescate de tumores resecados de forma incompleta tienen un desenlace inferior en comparación con los pacientes que se someten a trasplante como tratamiento quirúrgico primario.[Nivel de evidencia C1] Es fundamental realizar la cirugía adecuada en el momento de la resección.
El abordaje que usa el Children's Oncology Group (COG) en los ensayos clínicos de América del Norte es llevar a cabo una cirugía al inicio cuando se puede lograr una resección completa con una hemihepatectomía simple en la que se obtengan márgenes sin compromiso tumoral. En el ensayo AHEP0731 (NCT00980460) del COG se analizó el uso de PRETEXT y POSTEXT para determinar el abordaje quirúrgico óptimo y el momento oportuno de la cirugía. La agrupación por imágenes POSTTEXT se realizó después de 2 y 4 ciclos de quimioterapia para determinar el momento óptimo para la cirugía definitiva. Para obtener más información, consultar la sección Estratificación tumoral por imágenes y estadificación quirúrgica de Evans para el cáncer de hígado infantil.
Trasplante ortotópico de hígado
Los trasplantes de hígado son muy útiles para el tratamiento de niños con tumores hepáticos irresecables.; [Nivel de evidencia C1] En una revisión de la experiencia mundial se documentó una tasa de supervivencia posterior al trasplante de un 70 % a un 80 % para niños con hepatoblastomas. La invasión vascular intravenosa, el compromiso ganglionar y la diseminación extrahepática adyacente, no tuvieron un efecto adverso importante en los desenlaces. Es posible que la quimioterapia adyuvante después del trasplante disminuya el riesgo de recidiva tumoral, pero su uso no se ha estudiado de manera definitiva en un ensayo clínico aleatorizado.
Evidencia (trasplante ortotópico de hígado):
- Se consultó la base de datos United Network for Organ Sharing (UNOS) para obtener una lista de todos los pacientes menores de 18 años con un tumor hepático primario maligno sometidos a trasplante ortotópico de hígado entre 1987 y 2012 (N = 544). Los pacientes tenían un diagnóstico de hepatoblastoma (n = 376, 70 %), carcinoma hepatocelular (n = 84, 15 %) y otros tipos de tumores (n = 84, 15 %). Los pacientes con carcinoma hepatocelular eran mayores, fue más frecuente que estuvieran hospitalizados en el momento del trasplante y fue más probable que hubieran recibido un órgano cadavérico que los pacientes con hepatoblastoma.
- La tasa de supervivencia a 5 años de los pacientes fue del 73 % y la tasa de supervivencia del injerto fue del 74 % para toda la cohorte; la mayoría de las muertes fueron por neoplasia maligna. En el análisis multivariante, los factores pronósticos independientes para la supervivencia a 5 años del paciente y del injerto fueron los siguientes:
- Diagnóstico.
- Durante el período de estudio entre 1987 y 2012, en los pacientes con hepatoblastoma, la tasa de supervivencia a 5 años fue del 76 % y la tasa de supervivencia del injerto fue del 77 %; mientras que en los pacientes con carcinoma hepatocelular, la tasa de supervivencia de los pacientes y del injerto fueron ambas del 63 %.
- Durante el período de estudio entre 2009 y 2012, en los pacientes con hepatoblastoma, la tasa de supervivencia a 3 años y la tasa de supervivencia del injerto fueron del 84 %; mientras que en los pacientes con carcinoma hepatocelular, la tasa de supervivencia de los pacientes y del injerto fueron ambas del 85 %.
- Época del trasplante.
- La tasa de mortalidad según el cociente de riesgos instantáneos fue de 1,0 para el período anterior a 2002, de 0,72 para el período entre 2002 y 2009, y de 0,54 para el período entre 2009 y 2012.
- Estado clínico en el momento del trasplante.
- En los pacientes de hepatoblastoma, la tasa de supervivencia según el cociente de riesgos instantáneos fue de 1,0 en los pacientes hospitalizados versus 1,81 en los pacientes que no estaban hospitalizados en el momento del trasplante.
- En los pacientes con carcinoma hepatocelular, la tasa de supervivencia según el cociente de riesgos instantáneos fue de 1,0 en los pacientes hospitalizados versus 1,92 en los pacientes no hospitalizados.
- Los pacientes hospitalizados en la unidad de cuidados intensivos no evolucionaron de modo más precario que los pacientes que no estaban en esta unidad.
- Diagnóstico.
- La tasa de supervivencia a 5 años de los pacientes fue del 73 % y la tasa de supervivencia del injerto fue del 74 % para toda la cohorte; la mayoría de las muertes fueron por neoplasia maligna. En el análisis multivariante, los factores pronósticos independientes para la supervivencia a 5 años del paciente y del injerto fueron los siguientes:
- En un informe de 149 pacientes menores de 21 años con carcinoma hepatocelular que se sometieron a trasplantes entre 1987 y 2015, se utilizaron datos detallados recopilados por el U.S. Scientific Registry of Transplant Receptors en todos los centros de trasplantes pediátricos de los Estados Unidos.
- La tasa de supervivencia del injerto a 1 año fue de casi el 85 %, y no fue diferente a la de los pacientes con hepatoblastoma o atresia biliar. La supervivencia continuó descendiendo con el paso del tiempo, del 85 % a 1 año, al 52 % a 5 años y al 43 % a 10 años, una disminución mucho más drástica que la observada en el hepatoblastoma o la atresia biliar.
- La supervivencia posterior al trasplante no fue diferente a la de los adultos sometidos a trasplante por un carcinoma hepatocelular.
- De los pacientes con carcinoma hepatocelular, 22 recibieron el diagnóstico de carcinoma hepatocelular después de un trasplante por una enfermedad cirrótica, como la tirosinemia. Estos pacientes tuvieron un desenlace superior, pero no fue estadísticamente significativo en comparación con el resto de los 149 pacientes.
- En una revisión de la base de datos del Programa Surveillance, Epidemiology, and End Results (SEER) y de muchas series de instituciones individuales, se notificaron resultados similares al estudio de la base de datos de UNOS descrito antes.; [Nivel de evidencia C1]
- En un estudio de 3 instituciones de niños con carcinoma hepatocelular, la tasa general de supervivencia sin enfermedad a 5 años fue de alrededor del 60 %.
- En un estudio en el que se utilizó la base de datos Society of Pediatric Liver Transplantation (SPLIT) para identificar a los pacientes que se sometieron a un trasplante entre 2011 y 2019, se notificó lo siguiente:[Nivel de evidencia C2]
- La tasa de SSC a 3 años fue del 81 % en los pacientes con hepatoblastoma que recibieron un trasplante (n = 157).
- La tasa de SSC a 3 años fue del 62 % en los pacientes con carcinoma hepatocelular que recibieron un trasplante (n = 18).
- De los pacientes que recibieron un trasplante para tratar el hepatoblastoma, un 6,9 % tenían enfermedad PRETEXT II y un 15,3 % tenían enfermedad POSTTEXT I o II.
- La extensión tumoral no afectó la supervivencia (P = NS).
- Los pacientes que recibieron trasplantes para rescate (n = 13) y los pacientes que recibieron trasplantes para hepatoblastoma primario tuvieron tasas similares de SSC a 3 años (62 % vs. 78 %; P = NS).
- Entre los pacientes que recibieron trasplantes por carcinoma hepatocelular, la tasa de SSC a 3 años fue más precaria en los pacientes de edad avanzada (38 % en los pacientes ≥8 años vs. 86 % en los pacientes <8 años; P< 0,001).
La aplicación de los criterios de Milán para la selección hecha por la UNOS de los receptores de hígados de donantes fallecidos es objeto de controversia. Los criterios de Milán para el trasplante de hígado se aplican a adultos con cirrosis y carcinoma hepatocelular. Estos criterios no son útiles para niños y adolescentes con carcinoma hepatocelular; sobre todo, en aquellos sin cirrosis.
La cirrosis es un factor de riesgo subyacente para el carcinoma hepatocelular en niños que presentan ciertas enfermedades o afecciones. Estas enfermedades incluyen, entre otras, la hepatitis B perinatal, la tirosinemia hepatorrenal, la colestasis intrahepática familiar progresiva, una glucogenosis y el síndrome de Alagille. Las mejoras en la metodología de detección han permitido identificar y tratar de forma temprana algunas de estas afecciones, así como hacer el seguimiento para detectar el carcinoma hepatocelular. Sin embargo, debido a lo precario del pronóstico de los pacientes con carcinoma hepatocelular, se debe considerar el trasplante de hígado cuando el paciente tiene enfermedades o afecciones asociadas con hallazgos tempranos de cirrosis, antes de que se presente una insuficiencia hepática o una neoplasia maligna en el hígado.
El trasplante de hígado de donante vivo como tratamiento de una neoplasia maligna de hígado es más común durante la niñez y el desenlace es similar al de un trasplante de hígado cadavérico. En los pacientes con carcinoma hepatocelular, la invasión vascular macroscópica, las metástasis a distancia, el compromiso ganglionar, el tamaño del tumor y el sexo masculino fueron factores significativos de riesgo de recidiva. En un informe, 33 pacientes con hepatoblastoma y 10 pacientes con carcinoma hepatocelular se trataron con trasplantes de hígado de donante vivo. En los pacientes con hepatoblastoma, la tasa de SG a 5 años fue del 87,4 % y la tasa de supervivencia sin complicaciones (SSC) fue del 75,8 %. Las tasas de SG y SSC a 5 años fueron del 75,4 % en los pacientes con carcinoma hepatocelular. La presencia de invasión de la vena renal se relacionó con un aumento de la incidencia de recidiva y muerte (P = 0,28).[Nivel de evidencia C1]
Resección quirúrgica para la enfermedad metastásica
A menudo, se recomienda la resección quirúrgica para la enfermedad metastásica, pero la tasa de cura en niños con hepatoblastoma no se ha determinado por completo. La resección de las metástasis se puede hacer en áreas de enfermedad localmente invasiva (por ejemplo, diafragma) y en metástasis encefálicas aisladas. La resección de las metástasis pulmonares se debe considerar si el número de metástasis es bajo. En un estudio realizado en los Estados Unidos de 38 pacientes que tenían metástasis pulmonares en el momento del diagnóstico, solo 9 pacientes se sometieron a resección quirúrgica. El momento de la resección pulmonar en relación con la resección definitiva del tumor primario varió (2 pacientes antes, 5 pacientes de modo simultáneo y 2 pacientes después de la resección primaria). De los 9 pacientes, 8 sobrevivieron. De 20 niños con recaída restringida a los pulmones, todos los pacientes recibieron quimioterapia de rescate, 8 pacientes se sometieron a toracotomía y metastasectomía pulmonar, y 5 pacientes se sometieron a toracotomía y biopsia. De los 13 pacientes que se sometieron a cirugía, solo 4 sobrevivieron a largo plazo, 2 de ellos presentaban enfermedad en estadio I, y 2 enfermedad en estadio IV.
La ablación por radiofrecuencia también se ha utilizado para tratar el hepatoblastoma oligometastático cuando los pacientes prefieren evitar la metastasectomía quirúrgica.[Nivel de evidencia C1]
Quimioterapia
Los regímenes quimioterapéuticos utilizados para el tratamiento del hepatoblastoma y el carcinoma hepatocelular se describen en sus respectivas secciones. La quimioterapia ha tenido mucho más éxito en el tratamiento del hepatoblastoma que en el tratamiento del carcinoma hepatocelular. Para obtener más información, consultar las secciones Tratamiento del hepatoblastoma y Tratamiento del carcinoma hepatocelular.
El estándar de atención en los Estados Unidos es la quimioterapia preoperatoria cuando el tumor es irresecable y la quimioterapia posoperatoria después de la resección completa, incluso cuando se haya administrado quimioterapia preoperatoria. Se ha demostrado que el tratamiento con quimioterapia preoperatoria beneficia a los niños con hepatoblastoma; sin embargo, el uso de quimioterapia posoperatoria después de la resección quirúrgica definitiva o un trasplante de hígado no se ha investigado en estudios aleatorizados.
Radioterapia
La radioterapia, incluso en combinación con la quimioterapia, no ha curado a niños con tumores hepáticos irresecables. Sin embargo, en un estudio de 154 pacientes con hepatoblastoma, se observó que algunos pacientes tal vez no necesiten radioterapia o una segunda resección de márgenes con compromiso tumoral incluso cuando la resección del hepatoblastoma haya sido incompleta y se observe tumor residual microscópico. Aunque no hay una indicación estándar, la radioterapia quizás desempeñe una función en el tratamiento de los pacientes con hepatoblastomas que se resecaron de forma incompleta. La radioterapia corporal estereotáctica es una alternativa de tratamiento segura y eficaz que se ha utilizado con éxito en pacientes adultos con carcinoma hepatocelular que no pueden someterse a ablación o resección hepática. Este método de radioterapia altamente conformada, cuando está disponible, se puede considerar de manera individual en niños con carcinoma hepatocelular.
Otros abordajes de tratamiento
Los siguientes son otros abordajes de tratamiento:
- Quimioembolización transarterial (QETA). La QETA es un procedimiento no quirúrgico, mínimamente invasivo, guiado por imágenes, que se utiliza para el tratamiento de lesiones malignas en el hígado. Durante el procedimiento se utiliza un catéter para administrar medicamentos de quimioterapia y materiales de embolización en los vasos sanguíneos que irrigan el tumor. La ruta del catéter arterial es guiada por imágenes, con mayor frecuencia se pasa por la arteria hepática y, en ocasiones, antes de la inyección terapéutica se confirma en imágenes la irrigación sanguínea del tumor por la arteria que se va a tratar. Este procedimiento permite el tratamiento de tumores inaccesibles mediante cirugía convencional o tratamientos de radiación. La QETA se ha utilizado para pacientes con hepatoblastoma inoperable. Este procedimiento también se ha utilizado con éxito en algunos niños para achicar los tumores y permitir su resección.
- Radioembolización transarterial (RETA). La RETA es un procedimiento no quirúrgico, mínimamente invasivo y guiado por imágenes en el que se administra radioterapia para el tratamiento de tumores en el hígado. Con este procedimiento se coloran microesferas radioactivas y se bloquea el flujo arterial del tumor para mantener la radiación en el interior de este. Las microesferas de vidrio o resina, recubiertas por lo general con itrio Y 90 (90Y), se colocan dentro del tumor a través de catéteres insertados en las arterias que irrigan el tumor. Por lo común, se utiliza la arteria hepática o sus ramas, pero es posible que la irrigación del tumor provenga de manera parcial de vasos adyacentes parasitarios. Debido al riesgo de administrar radiación al pulmón vecino, se realizan imágenes con microagregados de albúmina con tecnecio Tc 99m que se inyectan por el catéter colocado antes de la administración de las microesferas radiactivas y de esta manera se mide rigurosamente la exposición del pulmón a la radiación. Si se determina que la exposición es peligrosa, no se administra RETA. La RETA con 90Y se ha usado en niños con hepatoblastoma (n = 2) y carcinoma hepatocelular (n = 2) que tienen tumores irresecables. Después del tratamiento con TARA 90Y, todos los tumores se resecaron por completo.[Nivel de evidencia C3]; [Nivel de evidencia C2] Este abordaje también se ha utilizado como paliación en niños con carcinoma hepatocelular. Para obtener más información, consultar Tratamiento del cáncer primario de hígado.
- Ultrasonidos focalizados de alta intensidad (HIFU). El HIFU es un tratamiento no invasivo para una amplia gama de tumores y enfermedades. Para el HIFU se usa un transductor de ecografía, similar a los que se usan para los estudios con imágenes, pero con mucha más energía. El transductor enfoca las ondas sonoras para generar calor en un solo punto dentro del cuerpo y destruir el tejido objetivo. El tejido se puede calentar hasta 66°C en solo 20 segundos. Este proceso se repite tantas veces como sea necesario hasta que se destruye el tejido objetivo. Las imágenes por resonancia magnética se utilizan para planificar el tratamiento y controlar la cantidad de calor en tiempo real. Una combinación de quimioterapia seguida de QETA e HIFU mostró resultados prometedores en China para los niños con tumores malignos de hígado PRETEXT III y PRETEXT IV, algunos de los cuales tenían tumores resecables pero no se sometieron a cirugía debido a negativa de los padres.
References
- Tiao GM, Bobey N, Allen S, et al.: The current management of hepatoblastoma: a combination of chemotherapy, conventional resection, and liver transplantation. J Pediatr 146 (2): 204-11, 2005.
- Czauderna P, Otte JB, Aronson DC, et al.: Guidelines for surgical treatment of hepatoblastoma in the modern era--recommendations from the Childhood Liver Tumour Strategy Group of the International Society of Paediatric Oncology (SIOPEL). Eur J Cancer 41 (7): 1031-6, 2005.
- Czauderna P, Mackinlay G, Perilongo G, et al.: Hepatocellular carcinoma in children: results of the first prospective study of the International Society of Pediatric Oncology group. J Clin Oncol 20 (12): 2798-804, 2002.
- Meyers RL, Czauderna P, Otte JB: Surgical treatment of hepatoblastoma. Pediatr Blood Cancer 59 (5): 800-8, 2012.
- Aronson DC, Meyers RL: Malignant tumors of the liver in children. Semin Pediatr Surg 25 (5): 265-275, 2016.
- Murawski M, Weeda VB, Maibach R, et al.: Hepatocellular Carcinoma in Children: Does Modified Platinum- and Doxorubicin-Based Chemotherapy Increase Tumor Resectability and Change Outcome? Lessons Learned From the SIOPEL 2 and 3 Studies. J Clin Oncol 34 (10): 1050-6, 2016.
- Allan BJ, Wang B, Davis JS, et al.: A review of 218 pediatric cases of hepatocellular carcinoma. J Pediatr Surg 49 (1): 166-71; discussion 171, 2014.
- Becker K, Furch C, Schmid I, et al.: Impact of postoperative complications on overall survival of patients with hepatoblastoma. Pediatr Blood Cancer 62 (1): 24-8, 2015.
- D'Antiga L, Vallortigara F, Cillo U, et al.: Features predicting unresectability in hepatoblastoma. Cancer 110 (5): 1050-8, 2007.
- Lautz TB, Ben-Ami T, Tantemsapya N, et al.: Successful nontransplant resection of POST-TEXT III and IV hepatoblastoma. Cancer 117 (9): 1976-83, 2011.
- Fonseca A, Gupta A, Shaikh F, et al.: Extreme hepatic resections for the treatment of advanced hepatoblastoma: Are planned close margins an acceptable approach? Pediatr Blood Cancer 65 (2): , 2018.
- Fuchs J, Cavdar S, Blumenstock G, et al.: POST-TEXT III and IV Hepatoblastoma: Extended Hepatic Resection Avoids Liver Transplantation in Selected Cases. Ann Surg 266 (2): 318-323, 2017.
- Baertschiger RM, Ozsahin H, Rougemont AL, et al.: Cure of multifocal panhepatic hepatoblastoma: is liver transplantation always necessary? J Pediatr Surg 45 (5): 1030-6, 2010.
- Felsted AE, Shi Y, Masand PM, et al.: Intraoperative ultrasound for liver tumor resection in children. J Surg Res 198 (2): 418-23, 2015.
- Rossi G, Tarasconi A, Baiocchi G, et al.: Fluorescence guided surgery in liver tumors: applications and advantages. Acta Biomed 89 (9-S): 135-140, 2018.
- Shen Q, Liu X, Pan S, et al.: Effectiveness of indocyanine green fluorescence imaging in resection of hepatoblastoma. Pediatr Surg Int 39 (1): 181, 2023.
- Otte JB, Pritchard J, Aronson DC, et al.: Liver transplantation for hepatoblastoma: results from the International Society of Pediatric Oncology (SIOP) study SIOPEL-1 and review of the world experience. Pediatr Blood Cancer 42 (1): 74-83, 2004.
- Venkatramani R, Stein JE, Sapra A, et al.: Effect of neoadjuvant chemotherapy on resectability of stage III and IV hepatoblastoma. Br J Surg 102 (1): 108-13, 2015.
- Vinayak R, Cruz RJ, Ranganathan S, et al.: Pediatric liver transplantation for hepatocellular cancer and rare liver malignancies: US multicenter and single-center experience (1981-2015). Liver Transpl 23 (12): 1577-1588, 2017.
- Guiteau JJ, Cotton RT, Karpen SJ, et al.: Pediatric liver transplantation for primary malignant liver tumors with a focus on hepatic epithelioid hemangioendothelioma: the UNOS experience. Pediatr Transplant 14 (3): 326-31, 2010.
- Malek MM, Shah SR, Atri P, et al.: Review of outcomes of primary liver cancers in children: our institutional experience with resection and transplantation. Surgery 148 (4): 778-82; discussion 782-4, 2010.
- Héry G, Franchi-Abella S, Habes D, et al.: Initial liver transplantation for unresectable hepatoblastoma after chemotherapy. Pediatr Blood Cancer 57 (7): 1270-5, 2011.
- Suh MY, Wang K, Gutweiler JR, et al.: Safety of minimal immunosuppression in liver transplantation for hepatoblastoma. J Pediatr Surg 43 (6): 1148-52, 2008.
- Zsíros J, Maibach R, Shafford E, et al.: Successful treatment of childhood high-risk hepatoblastoma with dose-intensive multiagent chemotherapy and surgery: final results of the SIOPEL-3HR study. J Clin Oncol 28 (15): 2584-90, 2010.
- Khan AS, Brecklin B, Vachharajani N, et al.: Liver Transplantation for Malignant Primary Pediatric Hepatic Tumors. J Am Coll Surg 225 (1): 103-113, 2017.
- Browne M, Sher D, Grant D, et al.: Survival after liver transplantation for hepatoblastoma: a 2-center experience. J Pediatr Surg 43 (11): 1973-81, 2008.
- Hamilton EC, Balogh J, Nguyen DT, et al.: Liver transplantation for primary hepatic malignancies of childhood: The UNOS experience. J Pediatr Surg : , 2017.
- McAteer JP, Goldin AB, Healey PJ, et al.: Surgical treatment of primary liver tumors in children: outcomes analysis of resection and transplantation in the SEER database. Pediatr Transplant 17 (8): 744-50, 2013.
- Reyes JD, Carr B, Dvorchik I, et al.: Liver transplantation and chemotherapy for hepatoblastoma and hepatocellular cancer in childhood and adolescence. J Pediatr 136 (6): 795-804, 2000.
- Boster JM, Superina R, Mazariegos GV, et al.: Predictors of survival following liver transplantation for pediatric hepatoblastoma and hepatocellular carcinoma: Experience from the Society of Pediatric Liver Transplantation (SPLIT). Am J Transplant 22 (5): 1396-1408, 2022.
- Otte JB: Should the selection of children with hepatocellular carcinoma be based on Milan criteria? Pediatr Transplant 12 (1): 1-3, 2008.
- de Ville de Goyet J, Meyers RL, Tiao GM, et al.: Beyond the Milan criteria for liver transplantation in children with hepatic tumours. Lancet Gastroenterol Hepatol 2 (6): 456-462, 2017.
- Khanna R, Verma SK: Pediatric hepatocellular carcinoma. World J Gastroenterol 24 (35): 3980-3999, 2018.
- Sevmis S, Karakayali H, Ozçay F, et al.: Liver transplantation for hepatocellular carcinoma in children. Pediatr Transplant 12 (1): 52-6, 2008.
- Faraj W, Dar F, Marangoni G, et al.: Liver transplantation for hepatoblastoma. Liver Transpl 14 (11): 1614-9, 2008.
- Pire A, Tambucci R, De Magnée C, et al.: Living donor liver transplantation for hepatic malignancies in children. Pediatr Transplant 25 (7): e14047, 2021.
- Feusner JH, Krailo MD, Haas JE, et al.: Treatment of pulmonary metastases of initial stage I hepatoblastoma in childhood. Report from the Childrens Cancer Group. Cancer 71 (3): 859-64, 1993.
- Zsiros J, Brugieres L, Brock P, et al.: Dose-dense cisplatin-based chemotherapy and surgery for children with high-risk hepatoblastoma (SIOPEL-4): a prospective, single-arm, feasibility study. Lancet Oncol 14 (9): 834-42, 2013.
- Meyers RL, Katzenstein HM, Krailo M, et al.: Surgical resection of pulmonary metastatic lesions in children with hepatoblastoma. J Pediatr Surg 42 (12): 2050-6, 2007.
- O'Neill AF, Towbin AJ, Krailo MD, et al.: Characterization of Pulmonary Metastases in Children With Hepatoblastoma Treated on Children's Oncology Group Protocol AHEP0731 (The Treatment of Children With All Stages of Hepatoblastoma): A Report From the Children's Oncology Group. J Clin Oncol 35 (30): 3465-3473, 2017.
- Yevich S, Calandri M, Gravel G, et al.: Reiterative Radiofrequency Ablation in the Management of Pediatric Patients with Hepatoblastoma Metastases to the Lung, Liver, or Bone. Cardiovasc Intervent Radiol 42 (1): 41-47, 2019.
- Weeda VB, Murawski M, McCabe AJ, et al.: Fibrolamellar variant of hepatocellular carcinoma does not have a better survival than conventional hepatocellular carcinoma--results and treatment recommendations from the Childhood Liver Tumour Strategy Group (SIOPEL) experience. Eur J Cancer 49 (12): 2698-704, 2013.
- Czauderna P, Lopez-Terrada D, Hiyama E, et al.: Hepatoblastoma state of the art: pathology, genetics, risk stratification, and chemotherapy. Curr Opin Pediatr 26 (1): 19-28, 2014.
- Schnater JM, Aronson DC, Plaschkes J, et al.: Surgical view of the treatment of patients with hepatoblastoma: results from the first prospective trial of the International Society of Pediatric Oncology Liver Tumor Study Group. Cancer 94 (4): 1111-20, 2002.
- Habrand JL, Nehme D, Kalifa C, et al.: Is there a place for radiation therapy in the management of hepatoblastomas and hepatocellular carcinomas in children? Int J Radiat Oncol Biol Phys 23 (3): 525-31, 1992.
- Wang PM, Chung NN, Hsu WC, et al.: Stereotactic body radiation therapy in hepatocellular carcinoma: Optimal treatment strategies based on liver segmentation and functional hepatic reserve. Rep Pract Oncol Radiother 20 (6): 417-24, 2015 Nov-Dec.
- Xianliang H, Jianhong L, Xuewu J, et al.: Cure of hepatoblastoma with transcatheter arterial chemoembolization. J Pediatr Hematol Oncol 26 (1): 60-3, 2004.
- Malogolowkin MH, Stanley P, Steele DA, et al.: Feasibility and toxicity of chemoembolization for children with liver tumors. J Clin Oncol 18 (6): 1279-84, 2000.
- Hirakawa M, Nishie A, Asayama Y, et al.: Efficacy of preoperative transcatheter arterial chemoembolization combined with systemic chemotherapy for treatment of unresectable hepatoblastoma in children. Jpn J Radiol 32 (9): 529-36, 2014.
- Aguado A, Dunn SP, Averill LW, et al.: Successful use of transarterial radioembolization with yttrium-90 (TARE-Y90) in two children with hepatoblastoma. Pediatr Blood Cancer 67 (9): e28421, 2020.
- Whitlock RS, Loo C, Patel K, et al.: Transarterial Radioembolization Treatment as a Bridge to Surgical Resection in Pediatric Hepatocellular Carcinoma. J Pediatr Hematol Oncol 43 (8): e1181-e1185, 2021.
- Hawkins CM, Kukreja K, Geller JI, et al.: Radioembolisation for treatment of pediatric hepatocellular carcinoma. Pediatr Radiol 43 (7): 876-81, 2013.
- Wang S, Yang C, Zhang J, et al.: First experience of high-intensity focused ultrasound combined with transcatheter arterial embolization as local control for hepatoblastoma. Hepatology 59 (1): 170-7, 2014.
Consideraciones especiales para el tratamiento de niños con cáncer
El cáncer en niños y adolescentes es infrecuente, aunque desde 1975 se ha observado un aumento gradual de la incidencia general. Los niños y adolescentes con cáncer se deben derivar a centros médicos que cuenten con equipos multidisciplinarios de especialistas en oncología con experiencia en el tratamiento de los cánceres infantiles. Este equipo multidisciplinario incorpora la pericia de los siguientes profesionales de atención de la salud y otros para asegurar que los niños reciban el tratamiento, los cuidados médicos de apoyo y la rehabilitación que les permitan lograr una supervivencia y calidad de vida óptimas:
- Médicos de atención primaria.
- Cirujanos pediatras y cirujanos especializados en trasplantes.
- Radioncólogos.
- Oncólogos o hematólogos pediatras.
- Especialistas en rehabilitación.
- Enfermeros especializados en pediatría.
- Trabajadores sociales.
- Profesionales de la vida infantil.
- Psicólogos.
- Nutricionistas.
Para obtener información específica sobre los cuidados médicos de apoyo para niños y adolescentes con cáncer, consultar los resúmenes de Cuidados médicos de apoyo y cuidados paliativos.
La American Academy of Pediatrics estableció pautas para los centros de oncología pediátrica y su función en el tratamiento de los pacientes de cáncer infantil. En estos centros de oncología pediátrica, se dispone de ensayos clínicos para la mayoría de los tipos de cáncer que se presentan en niños y adolescentes, y se ofrece la oportunidad de participar a la mayoría de los pacientes y familiares. Por lo general, los ensayos clínicos para los niños y adolescentes con cáncer se diseñan a fin de comparar un tratamiento que parece mejor con el tratamiento estándar actual. La mayoría de los avances en la identificación de tratamientos curativos para los cánceres infantiles se lograron mediante ensayos clínicos. Para obtener más información sobre ensayos clínicos en curso, consultar el portal de Internet del NCI.
Se han logrado mejoras notables en la supervivencia de niños y adolescentes con cáncer. Entre 1975 y 2020, la mortalidad por cáncer infantil disminuyó en más del 50 %. Los niños y adolescentes sobrevivientes de cáncer necesitan un seguimiento minucioso, ya que es posible que los efectos secundarios del tratamiento del cáncer persistan o se presenten meses o años después de este. Para obtener información específica sobre la incidencia, el tipo y la vigilancia de los efectos tardíos en los niños y adolescentes sobrevivientes de cáncer, consultar Efectos tardíos del tratamiento anticanceroso en la niñez.
References
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed December 15, 2023.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 15, 2023.
- Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed March 6, 2024.
Hepatoblastoma
Incidencia
La incidencia anual de hepatoblastoma en los Estados Unidos aumentó (más del doble), de 0,8 (1975–1983) a 2,0 (2018) casos por millón personas de 19 años o menos. Se desconoce la causa de este incremento, pero quizás contribuya el aumento de la supervivencia de lactantes prematuros con muy bajo peso al nacer, lo que se sabe que está relacionado con el hepatoblastoma. En Japón, el riesgo de hepatoblastoma en los niños que pesaron menos de 1000 g al nacer es 15 veces mayor que el riesgo en los niños con peso normal al nacer. Otros datos han confirmado la incidencia alta de hepatoblastoma en lactantes prematuros con peso muy bajo al nacer. Los intentos de identificar factores relacionados con el tratamiento de los lactantes nacidos prematuramente no han revelado ninguna causa indicativa del aumento de la incidencia de hepatoblastoma.
La edad de presentación del cáncer de hígado en la niñez se relaciona con el subtipo histológico del tumor. Por lo general, los hepatoblastomas se presentan antes de los 3 años de edad, y cerca del 90 % de los tumores malignos de hígado en niños de 4 años o menos son hepatoblastomas.
Factores de riesgo
Las afecciones relacionadas con un aumento del riesgo de hepatoblastoma se describen en el Cuadro 4.
| Trastornos relacionados | Hallazgos clínicos |
|---|---|
| Síndrome de Aicardi | Para obtener más información, consultar la sección Síndrome de Aicardi. |
| Síndrome de Beckwith-Wiedemann | Para obtener más información, consultar la sección Síndrome de Beckwith-Wiedemann y hemihiperplasia. |
| Poliposis adenomatosa familiar | Para obtener más información, consultar la sección Poliposis adenomatosa familiar. |
| Enfermedades por almacenamiento de glucógeno I–IV | Los síntomas varían según el trastorno. |
| Lactantes con bajo peso al nacer | Neonatos prematuros y neonatos pequeños para la edad gestacional. |
| Síndrome de Simpson-Golabi-Behmel | Macroglosia, macrosomía, anomalías renales y esqueléticas, y aumento de riesgo de tumor de Wilms. |
| Trisomía 18 y otras trisomías | Trisomía 18: Microcefalia y micrognatia, puños cerrados con dedos superpuestos y retraso del desarrollo. La mayoría de los pacientes (>90 %) mueren durante el primer año de vida. |
Síndrome de Aicardi
Se presume que el síndrome de Aicardi es una afección ligada al cromosoma X que se presenta exclusivamente en mujeres, lo que lleva a la hipótesis de que un gen mutado en el cromosoma X es mortal en varones. El síndrome se define, de manera clásica, como la presencia de agenesia del cuerpo calloso, laguna coriorretiniana y espasmos infantiles, con una facies característica. A menudo se encuentran otras alteraciones encefálicas, oculares y costovertebrales.
Síndrome de Beckwith-Wiedemann y hemihiperplasia
La incidencia de hepatoblastoma aumenta de 1000 a 10 000 veces en lactantes y niños con síndrome de Beckwith-Wiedemann. El riesgo de hepatoblastoma también aumenta en los pacientes con hemihiperplasia (antes llamada hemihipertrofia), una afección que produce asimetría entre el lado derecho e izquierdo del cuerpo cuando una parte del cuerpo crece más rápido de lo normal.
La causa más común del síndrome de Beckwith-Wiedemann son cambios epigenéticos y en este caso el síndrome es esporádico. En ocasiones, también obedece a mutaciones genéticas y en este caso el síndrome es familiar. Cualquiera de estos mecanismos se relaciona con un aumento de la incidencia de tumores embrionarios, como el tumor de Wilms y el hepatoblastoma. La expresión de ambos alelos de IGFR2 y el consiguiente aumento de la expresión del factor de crecimiento similar a la insulina 2 (IGF-2) se han relacionado con la macrosomía y los tumores embrionarios observados en pacientes con síndrome de Beckwith-Wiedemann. Los tipos de tumores embrionarios relacionados con el síndrome de Beckwith-Wiedemann esporádico a menudo exhiben cambios somáticos en el locus del síndrome de Beckwith-Wiedemann y en el IGF-2. Las características genéticas de los tumores en niños con hemihiperplasia no se han definido con claridad.
Para identificar neoplasias malignas abdominales en estadio temprano, todos los niños con síndrome de Beckwith-Wiedemann o hemihiperplasia aislada se someten a exámenes de detección regulares para muchos tipos de tumores mediante ecografía abdominal. La detección con pruebas de las concentraciones de alfafetoproteína (AFP) también ha sido muy útil para la detección temprana del hepatoblastoma en estos niños. Debido a que los hepatoblastomas que se descubren temprano son pequeños, el tratamiento puede reducir al mínimo el uso de la terapia adyuvante después de la cirugía. Sin embargo, en una recopilación cuidadosa de datos publicados sobre 1370 niños con síndrome de Beckwith-Wiedemann evaluado mediante epigenotipo se demostró que la prevalencia del hepatoblastoma fue del 4,7 % en aquellos con síndrome de Beckwith-Wiedemann causado por disomía uniparental paterna del cromosoma 11p15, de menos del 1 % en quienes tenían dos tipos de alteraciones en regiones de control de impronta, y ausente en quienes presentaban una mutación en CDKN1C. Los autores recomendaron que solo los niños con síndrome de Beckwith-Wiedemann causado por una disomía uniparental se sometieran a exámenes de detección del hepatoblastoma mediante ecografía abdominal y análisis de las concentraciones de AFP cada 3 meses desde los 3 meses hasta los 5 años de edad.
Poliposis adenomatosa familiar
Existe una relación entre el hepatoblastoma y la poliposis adenomatosa familiar (PAF). El riesgo de hepatoblastoma es 800 veces mayor en los niños de familias portadoras de una variante patógena del gen APC. La detección del hepatoblastoma en familias con PAF mediante ecografías y pruebas de las concentraciones de AFP es polémica porque se notificó que el hepatoblastoma se presenta en menos de un 1 % de los miembros de estas familias con PAF. Sin embargo, en un estudio de 50 niños con presunto hepatoblastoma esporádico evaluados de manera consecutiva, se notificó que 5 niños (10 %) tenían mutaciones de la línea germinal en APC.
La evidencia actual no permiten descartar la posibilidad de que la predisposición al hepatoblastoma se limite a un subconjunto específico de mutaciones en APC. En otro estudio de niños con hepatoblastoma, se observó un predominio de la mutación en la región 5' del gen, pero algunos pacientes presentaron mutaciones más cercanas a la región 3'. En este estudio preliminar se proporciona evidencia de que quizás sea apropiado hacer exámenes de detección de mutaciones en APC y cáncer de colon en niños con hepatoblastoma.
Los hepatoblastomas infantiles que no tienen mutaciones de la línea germinal en APC tampoco tienen mutaciones somáticas en el gen APC; sin embargo, a menudo los hepatoblastomas tienen mutaciones en el gen CTNNB1, cuya función está estrechamente relacionada con el gen APC.
Exámenes de detección para niños con predisposición al hepatoblastoma
En una publicación de la American Association for Cancer Research, se indicó que todos los niños con más de un 1 % de riesgo de hepatoblastoma se deben someter a exámenes de detección. Esto incluye a pacientes con síndrome de Beckwith-Wiedemann, hemihiperplasia, síndrome de Simpson-Golabi-Behmel y trisomía 18. Los exámenes de detección con ecografía abdominal y pruebas de AFP se hacen cada 3 meses desde el nacimiento (o diagnóstico) hasta los 4 años, lo que identificará entre el 90 % al 95 % de los hepatoblastomas que se presentan en estos niños.
Características genómicas del hepatoblastoma
Características moleculares del hepatoblastoma
Los hallazgos genómicos relacionados con el hepatoblastoma son los siguientes:
- La frecuencia de mutaciones en el hepatoblastoma, según lo determinaron 3 grupos mediante secuenciación del exoma completo, fue muy baja (cerca de 3 variantes por tumor) en niños menores de 5 años. En un estudio genómico de todos los tipos de cáncer en pediatría se encontró que el hepatoblastoma tuvo la tasa de mutaciones génicas más baja entre todos los tipos de cáncer infantil analizados.
- El hepatoblastoma es primariamente una enfermedad de la activación de la vía WNT. El principal mecanismo de activación de la vía WNT son las mutaciones activadoras o deleciones que comprometen el exón 3 de CTNNB1. Se notificaron mutaciones en CTNNB1 en más del 80 % de los casos. Una causa menos común de activación de la vía WNT en el hepatoblastoma son las mutaciones en APC, que se asocian con la poliposis adenomatosa familiar.
- Se identificaron mutaciones en NFE2L2 en 10 de 174 (6 %), 4 de 88 (5 %), y 5 de 112 (4 %) casos de hepatoblastoma. La presencia de mutaciones en NFE2L2 se asoció con una tasa más baja de supervivencia.
- De manera comparable, las mutaciones en NFE2L2 se han encontrado en muchos tipos de cáncer, como el carcinoma hepatocelular. Estas mutaciones hacen que NFE2L2 sea insensible a la degradación mediada por KEAP1, lo que lleva a la activación de la vía NFE2L2-KEAP1, que a su vez activa la resistencia al estrés oxidativo y se cree que confiere resistencia ante la quimioterapia.
- Las mutaciones en TERT y TP53, que son frecuentes en el carcinoma hepatocelular de adultos, son infrecuentes en el hepatoblastoma pediátrico. Los casos de hepatoblastoma con mutaciones en TERT se presentan a una edad significativamente mayor, en comparación con los casos de hepatoblastoma sin mutaciones en TERT (mediana de edad en el momento del diagnóstico, alrededor de 10 años vs. 1,4 años).
- La disomía uniparental en 11p15.5 con pérdida del alelo materno se notificó en 6 de 15 casos de hepatoblastoma. Este hallazgo se confirmó en los estudios de caracterización genómica, donde se observó un desequilibrio alélico en el locus 11p15 en un 30 % a un 40 % de los casos.
La expresión génica y el perfil epigenético se han usado para identificar los subtipos biológicos de hepatoblastoma y para evaluar la importancia pronóstica de cada uno.
- Una firma de expresión de 16 genes dividió los casos de hepatoblastoma en 2 subtipos: C1 y C2. El subtipo C1 abarcó la mayoría de los casos de tipo histológico fetal bien diferenciado (fetal puro). El subtipo C2 exhibió una configuración más inmadura y se asoció con tasas más altas de enfermedad metastásica en el momento del diagnóstico. En un estudio de 174 pacientes con hepatoblastoma, el subtipo C2 fue un factor de predicción significativo de desenlace precario en un análisis multivariante.
- Otro grupo de investigación también encontró que el perfil de expresión génica se puede usar para identificar subtipos de hepatoblastoma con pronóstico favorable versus pronóstico desfavorable. El grupo de pronóstico desfavorable mostró expresión elevada de genes asociados con las células madre embrionarias y células progenitoras (por ejemplo, LIN28B, SALL4 y HMGA2). El grupo de pronóstico favorable mostró expresión elevada de genes asociados con la diferenciación hepática (por ejemplo, HNF1A).
- Se identificó una firma de expresión génica del cromosoma 14q32 (por ejemplo, DLK1), con una señal de expresión más fuerte vinculada con un riesgo más alto de fracaso del tratamiento. Una firma de expresión 14q32 fuerte también se observó en tejido hepático fetal, lo que apoya más el concepto de que los casos de hepatoblastoma con características biológicas similares a las de las células precursoras hepáticas exhiben un pronóstico más precario.
- El perfil epigenético del hepatoblastoma se ha usado para identificar subtipos de hepatoblastoma definidos por características moleculares. Se evaluaron los tumores de 113 pacientes con hepatoblastoma usando matrices de metilación del DNA. Se identificaron dos subtipos diferenciados, los grupos epigenéticos A y B (Epi-CA y Epi-CB). El perfil de metilación del grupo Epi-CB se parece al perfil del tejido hepático en fases iniciales de desarrollo embrionario o fetal. El perfil de metilación del grupo Epi-CA fue similar al del tejido hepático en fases fetales tardías o después del nacimiento. La supervivencia sin complicaciones fue significativamente más baja en los pacientes con el subtipo Epi-CB que en los pacientes con el subtipo Epi-CA.
La delimitación de las aplicaciones clínicas de los métodos para obtener el perfil genómico, transcriptómico y epigenómico con el fin de clasificar el riesgo en pacientes con hepatoblastoma exige una validación independiente, que es uno de los objetivos del Paediatric Hepatic International Tumour Trial (PHITT [NCT03017326]).
Diagnóstico
Biopsia
Siempre se indica una biopsia para asegurar el diagnóstico de un tumor hepático en pediatría, excepto por alguna de las siguientes circunstancias:
- Hemangioma hepático infantil (lactantes). La biopsia no se indica en los lactantes con hemangioma hepático infantil cuando se observan hallazgos clásicos en las imágenes por resonancia magnética (IRM). Si el diagnóstico resulta dudoso luego de imágenes de alta calidad, solo entonces se confirma mediante biopsia.
- Hiperplasia nodular focal. Es posible que no se indique o que se pueda retrasar una biopsia en los pacientes con hiperplasia nodular focal que muestran características clásicas en las IRM cuando se usa un contraste específico para hepatocitos. Si el diagnóstico resulta dudoso, se confirma mediante biopsia.
- En las directrices quirúrgicas del Children's Oncology Group (COG) (apéndice de AHEP0731 [NCT00980460]), se recomienda la resección del tumor en el momento del diagnóstico sin quimioterapia preoperatoria para los niños con tumores del grupo I de PRE-Treatment EXTent of disease (PRETEXT) y tumores del grupo II de PRETEXT con márgenes radiográficos de más de 1 cm en la vena cava, las venas hepáticas medias y la vena porta. En consecuencia, no se suele recomendar una biopsia en estas circunstancias.
- Coriocarcinoma hepático infantil (de lactantes). A menudo se indica quimioterapia sin biopsia en lactantes con coriocarcinoma hepático infantil, que se puede diagnosticar mediante imágenes y concentraciones muy elevadas de gonadotropina coriónica humana (GCH-ß).
Marcadores tumorales
Los marcadores tumorales AFP y GCH-β son útiles para el diagnóstico y tratamiento de los tumores hepáticos. Si bien la AFP está elevada en la mayoría de los niños con neoplasias hepáticas malignas, no es patognomónica de un tumor hepático maligno. La concentración de AFP puede elevarse con un tumor benigno o un tumor sólido maligno. En neonatos es normal que la concentración de la AFP esté muy elevada sin que sea a causa de un tumor, dicha concentración disminuye de manera progresiva después del nacimiento. La semivida de la AFP es de 5 a 7 días, y alrededor del primer año de vida debe estar en el intervalo normal de menos de 10 ng/ml. Las concentraciones de GCH-ß a veces también están elevadas en el hepatoblastoma o carcinoma hepatocelular, lo que lleva a precocidad isosexual en los niños varones.
Pronóstico y factores pronósticos
Pronóstico
La tasa de supervivencia general (SG) a 5 años para los niños con hepatoblastoma es del 70 %. Los neonatos con hepatoblastoma tienen desenlaces comparables a los niños mayores de hasta 5 años de edad.
Un grupo de colaboración integrado por 4 grupos de estudio (International Childhood Liver Tumors Strategy Group [SIOPEL], Children's Oncology Group [COG], Gesellschaft für Pädiatrische Onkologie und Hämatologie [GPOH], y Japanese Study Group for Pediatric Liver Tumor [JPLT]), se denominó Children's Hepatic Tumor International Collaboration (CHIC). El grupo CHIC analizó la supervivencia en una base de datos colaborativa de 1605 pacientes con hepatoblastoma tratados en 8 ensayos clínicos multicéntricos separados, con revisión central de todas las imágenes tumorales y de la información histológica detallada. En todos los resultados del estudio internacional se incluyeron los pacientes sometidos a trasplante ortotópico de hígado.
Las tasas de supervivencia a 5 años, con independencia de los factores de anotación, fueron las siguientes:
- 90 % en pacientes con tumores del grupo PRETEXT I.
- 83 % en pacientes con tumores del grupo PRETEXT II.
- 73 % en pacientes con tumores del grupo PRETEXT III.
- 52 % en pacientes con tumores del grupo PRETEXT IV.
Cuando se examinó cada factor de anotación por separado, sin tener en cuenta el grupo PRETEXT u otros factores de anotación de cada paciente, las tasas de SG a 5 años fueron las siguientes:
- 51 % en pacientes con factor de anotación V (compromiso de las 3 venas hepáticas o de la vena cava inferior).
- 49 % en pacientes con factor de anotación P (compromiso de las venas portas derecha e izquierda).
- 53 % en pacientes con factor de anotación E (tumor extrahepático adyacente).
- 52 % en pacientes con factor de anotación F (compromiso multifocal).
- 51 % en pacientes con factor de anotación R (ruptura del tumor).
- 41 % en pacientes con factor de anotación M (metástasis a distancia).
Para obtener más información sobre la agrupación de PRETEXT y los factores de anotación, consultar la sección Definiciones de los grupos PRETEXT y POSTTEXT.
Pronóstico del hepatoblastoma según el estadio quirúrgico de Evans. Los protocolos de estudio actuales utilizan la estadificación PRETEXT para determinar el pronóstico. El pronóstico histórico, que se basaba en el estadio de Evans, se indica a continuación. Para obtener más información, consultar la sección Estadificación quirúrgica de Evans para el cáncer de hígado infantil (histórica).
- Estadios I y II.
Cerca del 20 % al 30 % de los niños con hepatoblastoma tienen una enfermedad en estadio l o II. El pronóstico varía según el subtipo de hepatoblastoma:
- Los pacientes con tumores de subtipo histológico fetal bien diferenciado (antes conocidos como fetales puros) (4 % de los hepatoblastomas) tienen una tasa de SG a 3 y 5 años de un 100 % con quimioterapia mínima o sin quimioterapia, ya sean de los grupos PRETEXT I, II o III.
- Los pacientes con hepatoblastomas de otro subtipo histológico diferente al fetal bien diferenciado o al indiferenciado de células pequeñas tienen una tasa de SG a 3 y 4 años de un 90 % a un 100 % con quimioterapia adyuvante.
- Si hay algún componente indiferenciado de células pequeñas en pacientes con hepatoblastoma en estadio I o II, la tasa de supervivencia a 3 años es del 40 % al 70 %.
- Estadio III.
Cerca del 50 % al 70 % de los niños con hepatoblastoma tienen una enfermedad en estadio III. La tasa de SG a 3 y 5 años para los niños con hepatoblastoma en estadio III es inferior al 70 %.
- Estadio IV.
Cerca del 10 % al 20 % de los niños con hepatoblastoma tienen una enfermedad en estadio IV. La tasa de SG a 3 y 5 años para los niños con hepatoblastoma en estadio IV varía bastante, desde un 20 % hasta cerca de un 60 %, según los informes publicados. El estadio IV posquirúrgico es equivalente a cualquier grupo PRETEXT con un factor de anotación M.
Factores pronósticos
Los grupo de estudio de cáncer infantil, de manera individual, han intentado definir la importancia relativa de varios factores pronósticos presentes en el momento del diagnóstico y en respuesta al tratamiento. El grupo de estudio CHIC combinó de forma retrospectiva los datos de 8 ensayos clínicos (n = 1605) realizados entre 1988 y 2010. Este grupo publicó un análisis univariado del efecto en la supervivencia sin complicaciones (SSC) de los factores clínicos de pronóstico presentes en el momento del diagnóstico. El análisis confirmó muchos de los factores adversos estadísticamente significativos que se describen a continuación:
- Grupo PRETEXT más alto.
- Presencia de los siguientes factores de anotación PRETEXT:
- V: compromiso de las 3 venas hepáticas o la vena cava intrahepática inferior.
- P: compromiso de las ramas portales izquierda y derecha.
- E: extensiones tumorales extrahepáticas adyacentes (por ejemplo, diafragma u órganos adyacentes).
- F: tumores multifocales.
- R: ruptura del tumor.
- M: metástasis a distancia; con frecuencia en el pulmón.
- Concentración baja de AFP (<100 ng/ml o 100–1000 ng/ml para incluir a los lactantes con concentraciones elevadas de AFP).
- Edad más avanzada. Los pacientes de 3 a 7 años tienen un desenlace más precario en el grupo PRETEXT IV. Los pacientes de 8 años o más tienen un desenlace más precario que los pacientes más jóvenes de todos los grupos PRETEXT. En un informe posterior del grupo CHIC, el riesgo de un episodio aumentó con la edad en todas las cohortes de edad.[Nivel de evidencia C1] El aumento de la edad atenuó el efecto de otros factores de riesgo, como las metástasis, la concentración de AFP inferior a 100 ng/ml, la ruptura tumoral y la presencia de un factor de anotación.
Por el contrario, en los estudios SIOPEL-2 y 3, los lactantes menores de 6 meses presentaron grupos de PRETEXT, factores de anotación y desenlaces similares a los de los niños mayores sometidos al mismo tratamiento.[Nivel de evidencia C1]
En el estudio CHIC, el sexo, la prematuridad, el peso al nacer y el síndrome de Beckwith-Wiedemann no tuvieron efecto en la SSC.
Se publicó un análisis multivariante de estos factores pronósticos para ayudar a formular una nueva clasificación de grupos de riesgo para el hepatoblastoma. Esta clasificación se utilizó para generar un esquema de estratificación del riesgo que se utilizará en ensayos clínicos internacionales. Para obtener más información, consultar la sección Modelo internacional de clasificación del riesgo.
En otros estudios se observaron los siguientes factores que afectan el pronóstico:
- Grupo PRETEXT. En los estudios de SIOPEL, presentar tumores de un grupo PRETEXT bajo en el momento del diagnóstico (tumores PRETEXT I, II y III) es un factor de pronóstico favorable, mientras que tener un grupo PRETEXT IV es un factor de pronóstico precario. Para obtener más información, consultar la sección Estratificación tumoral por imágenes y estadificación quirúrgica de Evans para el cáncer de hígado infantil.
- Estadio tumoral. En los estudios del COG, los pacientes con tumores en estadio l que se resecaron en el momento del diagnóstico y que presentaban las características histológicas habituales del hepatoblastoma tuvieron desenlaces favorables cuando se los trató con quimioterapia limitada. Los pacientes con tumores del subtipo histológico fetal bien diferenciado presentaron un pronóstico excelente. Estos tumores por lo general no se tratan con quimioterapia. Los pacientes con tumores de otros estadios y subtipos histológicos reciben un tratamiento más intensivo.
- Factores relacionados con el tratamiento:
Quimioterapia. La quimioterapia suele reducir el tamaño y la extensión del hepatoblastoma, lo que permite una resección completa. La respuesta favorable del tumor primario a la quimioterapia, definida como una disminución del 30 % del tamaño tumoral según los Response Evaluation Criteria In Solid Tumors (RECIST) o una disminución del 90 % o más en las concentraciones de la AFP, predijo la resecabilidad del tumor. A su vez, esta respuesta favorable predijo la SG en todos los grupos de riesgo CHIC tratados con quimioterapia neoadyuvante en el ensayo clínico nacional japonés JPLT-2.[Nivel de evidencia B4]
Cirugía. La cura del hepatoblastoma exige la resección macroscópica del tumor. La mayoría de los hepatoblastomas son unifocales, por lo tanto, la resección suele ser posible. Si se extirpa por completo un hepatoblastoma, la mayoría de los pacientes sobreviven. Sin embargo, debido a compromiso vascular o de otro tipo, menos de un tercio de los pacientes tienen lesiones susceptibles de resección completa en el momento del diagnóstico. Es de vital importancia que un niño con probable hepatoblastoma sea evaluado por un cirujano pediátrico con experiencia en los métodos de resección hepática extrema con reconstrucción vascular. El niño también debe tener acceso a un programa de trasplante de hígado. Para los tumores en estadio avanzado, el tratamiento quirúrgico del hepatoblastoma es un procedimiento complicado. Las complicaciones posoperatorias en los pacientes de riesgo alto disminuyen la tasa de SG.
Trasplante ortotópico de hígado. El trasplante ortotópico de hígado es otra opción de tratamiento para los pacientes cuyo tumor permanece irresecable después de la quimioterapia preoperatoria; sin embargo, la presencia de tumor residual microscópico en el margen quirúrgico no impide un desenlace favorable. Esto puede ser el resultado de los otros ciclos de quimioterapia que se administran antes o después de la resección.
Para obtener más información sobre los desenlaces relacionados con regímenes quimioterapéuticos específicos, consultar el Cuadro 6.
- Factores relacionados con los marcadores tumorales:
El 90 % de los niños con hepatoblastoma y dos tercios de los niños con carcinoma hepatocelular exhiben concentraciones elevadas del marcador tumoral sérico AFP; este marcador aumenta en forma paralela al grado de actividad de la enfermedad. Las concentraciones de AFP en el momento del diagnóstico y la tasa de disminución de estas durante el tratamiento se comparan con un intervalo normal ajustado por edad. La ausencia de una disminución significativa en las concentraciones de AFP durante el tratamiento en ocasiones predice una respuesta precaria al tratamiento. En un estudio exploratorio de 34 niños con hepatoblastoma, la tasa de disminución de la AFP y el volumen tumoral, pero no las mediciones RECIST I, después de 2 ciclos de tratamiento tras el diagnóstico fue predictiva de SSC y SG.
La ausencia de concentraciones elevadas de AFP en el momento del diagnóstico (AFP <100 ng/ml) se presenta en un porcentaje pequeño de niños con hepatoblastoma y parece vincularse con un pronóstico muy precario, del mismo modo que la variante de hepatoblastoma indiferenciado de células pequeñas. Algunas de estas variantes no expresan SMARCB1 y se pueden considerar tumores rabdoides de hígado, que requieren otro tipo de tratamiento. Todos los hepatoblastomas indiferenciados de células pequeñas se someten a pruebas inmunohistoquímicas para determinar la pérdida de expresión de SMARCB1 con el fin de determinar aquellos que se deben tratar como hepatoblastoma versus aquellos que se deben tratar como tumores rabdoides de hígado.
Las concentraciones de GCH-ß a veces también están elevadas en el hepatoblastoma o carcinoma hepatocelular, lo que lleva a precocidad isosexual en los niños varones.
- Subtipo histológico del tumor:
Para obtener más información, consultar la sección Características histológicas en Hepatoblastoma.
Se han indicado otras variables como factores de pronóstico precario, pero ha sido difícil definir la importancia relativa de su significación pronóstica. En el estudio SIOPEL-1, un análisis multivariante de factores pronósticos luego de una respuesta favorable a la quimioterapia, indicó que solo una variable, PRETEXT, predijo la SG, mientras que la presencia de metástasis y el grupo PRETEXT predijeron la SSC. En un análisis de orden logarítmico de un estudio intergrupal de los Estados Unidos que se hizo desde el momento del diagnóstico, los subtipos histológicos fetal bien diferenciado e indiferenciado de células pequeñas y las concentraciones de AFP menores de 100 ng/ml fueron pronósticos. El grupo PRETEXT fue pronóstico en los pacientes asignados al grupo III, pero no al grupo IV. En el estudio CHIC se incorporaron datos detallados de pacientes con hepatoblastoma de múltiples grupos, lo que estableció una base sólida de factores de riesgo.
Características histológicas
El hepatoblastoma surge de precursores de hepatocitos que pueden presentar diferente aspecto morfológico, como sigue:
- Células pequeñas sin diferenciación epitelial ni estromal. Es imprescindible diferenciar entre un hepatoblastoma indiferenciado de células pequeñas que expresa SMARCB1 y un tumor rabdoide de hígado, que no tiene el gen SMARCB1 ni expresa SMARCB1. Es posible que ambas enfermedades compartan características histológicas similares. En ocasiones se necesitan abordajes y quimioterapias diferentes para el tratamiento óptimo de un tumor rabdoide de hígado y de un hepatoblastoma indiferenciado de células pequeñas. Para obtener más información sobre las diferencias entre estas dos enfermedades, consultar la sección Hepatoblastoma de subtipo histológico indiferenciado de células pequeñas y tumores rabdoides de hígado.
- Células epiteliales embrionarias que se asemejan al epitelio hepático entre las 6 y 8 semanas de gestación.
- Hepatocitos fetales bien diferenciados que, desde el punto de vista morfológico son indistinguibles de las células hepáticas fetales normales.
La mayoría de las veces, el tumor se compone de una mezcla de precursores de hepatocitos epiteliales. Cerca del 20 % de los tumores contienen derivados estromales como elementos osteoides, condroides y rabdoides. En ocasiones, se encuentran elementos neuronales, melanocíticos, escamosos y enteroendocrinos. Los siguientes subtipos histológicos tienen importancia clínica:
- Hepatoblastoma de subtipo histológico fetal bien diferenciado (fetal puro).
- Hepatoblastoma de subtipo mixto con células epiteliales fetales y embrionarias.
- Hepatoblastoma de subtipo histológico indiferenciado de células pequeñas y tumores rabdoides de hígado.
- Hepatoblastoma indiferenciado de células pequeñas (positivo para SMARCB1).
- Tumor rabdoide de hígado (negativo para SMARCB1).
Hepatoblastoma de subtipo histológico fetal bien diferenciado (fetal puro)
En un análisis de pacientes con hepatoblastomas resecados al inicio (antes de recibir quimioterapia), se indicó que los pacientes con tumores bien diferenciados de subtipo histológico fetal (antes llamados fetales puros) tienen un pronóstico mejor que los pacientes con una mezcla de componentes embrionarios más primitivos y de división celular rápida, u otros tejidos indiferenciados. En los estudios se notificó lo siguiente.
- En un estudio de pacientes con hepatoblastoma y tumores de subtipo histológico fetal bien diferenciado, se observó lo siguiente:
- La tasa de supervivencia en pacientes que recibieron 4 dosis de doxorrubicina en monoterapia fue del 100 %. Esto indica que los pacientes con tumores de subtipo histológico fetal bien diferenciado tal vez no necesiten quimioterapia después de la resección completa.
- En un estudio del COG (COG-P9645), 16 pacientes con hepatoblastoma de subtipo histológico fetal bien diferenciado con 2 o menos mitosis por 10 campos de gran aumento no se trataron con quimioterapia. En retrospectiva, los grupos PRETEXT fueron el grupo I (n = 4), el grupo II (n = 6) y el grupo III (n = 2).
- La tasa de supervivencia fue del 100 % en estos pacientes que no recibieron quimioterapia.
- Los 16 pacientes que participaron estaban vivos sin indicios de enfermedad al cabo de una mediana de seguimiento de 4,9 años (intervalo, 9 meses a 9,2 años).
Por tanto, la resección completa de un hepatoblastoma de subtipo histológico fetal bien diferenciado quizás indique que no se necesita quimioterapia.
Hepatoblastoma de subtipo histológico indiferenciado de células pequeñas y tumores rabdoides de hígado
El hepatoblastoma indiferenciado de células pequeñas (que conserva SMARCB1) es una variante poco común del hepatoblastoma. Desde el punto de vista histológico, el hepatoblastoma indiferenciado de células pequeñas se tipifica por una población difusa de células pequeñas con poco citoplasma que parecen neuroblastos. En la actualidad se reconoce que el hepatoblastoma indiferenciado de células pequeñas puede ser difícil de diferenciar del tumor rabdoide maligno de hígado, que en estudios anteriores se combinó con el hepatoblastoma indiferenciado de células pequeñas.
El hepatoblastoma de subtipo histológico indiferenciado de células pequeñas y los tumores rabdoides de hígado se distinguen por las siguientes anomalías características:
- Anomalías cromosómicas. Estas anomalías en los tumores rabdoides incluyen translocaciones que afectan un punto de ruptura en el cromosoma 22q11 y la deleción homocigótica en la región del cromosoma 22q12 que alberga el gen SMARCB1.
- Ausencia de expresión de SMARCB1. La falta de detección de SMARCB1 en el análisis inmunohistoquímico es característica de los tumores rabdoides malignos.
Históricamente, se notificó que el hepatoblastoma indiferenciado de células pequeñas se presentaba a una edad más temprana (6–10 meses) que otros casos de hepatoblastoma y se relacionó con concentraciones de AFP normales para la edad en el momento de la presentación. Sin embargo, en un estudio prospectivo del Children's Oncology Group (AHEP0731 [NCT00980460]), la presencia de características histológicas indiferenciadas de células pequeñas no se correlacionó con la edad, el sexo o las concentraciones de AFP en el momento del diagnóstico.
En el ensayo en curso, Paediatric Hepatic International Tumour Trial (PHITT), se designa cualquier tumor hepático infantil como un tumor rabdoide de hígado si contiene células que carecen de la expresión de SMARCB1. Los pacientes con tumores negativos para SMARCB1, que se presume están relacionados con los tumores rabdoides, tal vez no se incluyan en el ensayo internacional en el que se aborda el tratamiento del hepatoblastoma que incluye el subtipo histológico indiferenciado de células pequeñas, el carcinoma hepatocelular y las neoplasias malignas hepáticas infantiles sin otra indicación (SAI), pero no el tumor rabdoide de hígado. En este ensayo, se requiere que todos los pacientes con características histológicas compatibles con hepatoblastoma indiferenciado de células pequeñas puro, según la evaluación del patólogo institucional, se sometan a pruebas del gen SMARCB1 por análisis inmunohistoquímico de acuerdo con las prácticas de la institución. Además, la presencia de componentes blastémicos indica que es un hepatoblastoma convencional. Para obtener más información sobre el ensayo AHEP1531, consultar la sección Opciones de tratamiento en evaluación clínica para el hepatoblastoma.
Una característica compartida por el hepatoblastoma indiferenciado de células pequeñas y el tumor rabdoide maligno es el pronóstico precario. Sin embargo, debido a que el hepatoblastoma indiferenciado de células pequeñas y el tumor rabdoide de hígado no se discriminaron en estudios anteriores, es posible que algunas de las características pronósticas atribuidas al primero sean en realidad atribuibles en parte al segundo. Los estudios publicados sobre características pronósticas del subtipo histológico indiferenciado de células pequeñas son los siguientes:
- En 2009, se notificaron los resultados de un estudio de 11 niños con concentraciones bajas de AFP y características morfológicas de células pequeñas. Entre estos niños, 10 murieron por progresión de la enfermedad y 1 murió por complicaciones. De los 6 niños sometidos a pruebas del gen, todos eran negativos para SMARCB1, pero solo 1 exhibía características morfológicas rabdoides. Estos hallazgos indican que muchos tumores de hígado, o quizás todos los tumores de hígado con características morfológicas de células pequeñas y concentraciones de AFP muy bajas en niños pequeños pueden ser tumores rabdoides hepáticos. Estos tumores tienen un pronóstico precario que se relaciona con la mutación oncoiniciadora.
- En un estudio de una sola institución de 7 niños con tumores de hígado con características morfológicas de células pequeñas, se encontró que todos conservaban la expresión de SMARCB1. Sobrevivieron 6 niños, 1 niño murió por complicaciones del trasplante de hígado.
- En un estudio de 23 tumores de hígado del banco de tumores de Kiel, se encontraron 12 tumores con características morfológicas de células pequeñas. Nueve tumores exhibían características histológicas rabdoides malignas clásicas y 2 tumores tenían características histológicas mixtas de células pequeñas y rabdoides. No se proporcionaron desenlaces, pero se observó que los tumores rabdoides en el encéfalo tenían características de células pequeñas, en lugar de rabdoides clásicas.
- En un estudio de una sola institución de 6 niños con tumores de hígado negativos para SMARCB1, murieron 2 niños con características morfológicas de células pequeñas. Los 4 niños restantes con características histológicas rabdoides clásicas no recibieron tratamiento a base de cisplatino; 3 niños sobrevivieron y 1 niño murió por complicaciones del trasplante.
- En un informe del ensayo del COG COG AHEP0731 (NCT00980460), se identificó a 35 de 177 pacientes evaluables (19 %) con hepatoblastoma indiferenciado de células pequeñas confirmado por la revisión central. Se conservó la expresión nuclear de SMARCB1 en 33 de 35 pacientes. A diferencia de los informes anteriores, la presencia de características histológicas indiferenciadas de células pequeñas no se correlacionó con la edad, el sexo o las concentraciones de AFP en el momento del diagnóstico. Las tasas de SSC a 5 años en los pacientes con hepatoblastoma indiferenciado de células pequeñas de riesgo bajo, intermedio y alto fueron del 86 % (IC 95 %, 33–98 %), 81 % (IC 95 %, 51–92 %) y 29 % (IC 95 %, 4–81 %), respectivamente. Las tasas de SSC a 5 años en los pacientes con hepatoblastoma de riesgo bajo, intermedio y alto sin características histológicas indiferenciadas de células pequeñas fueron del 87 % (IC 95 %, 72–95 %), del 88 % (IC 95 %, 79–95 %) y del 55 % (IC 95 %, 33–74 %); P = 0,17), respectivamente. En este ensayo, la concordancia entre la revisión local y la revisión central fue precaria y solo fue concordante en 9 de 35 casos (26 %). Se hizo análisis inmunohistoquímico de expresión de SMARCB1 en todos los tumores. En este estudio, el hepatoblastoma que de otro modo se consideraría de riesgo muy bajo o riesgo bajo se actualizó a riesgo intermedio si se encontraban elementos indiferenciados de células pequeñas. Para obtener más información, consultar el Cuadro 5
Es posible que los resultados del ensayo CHIC sobre tumores hepáticos infantiles aclaren algunos de los aspectos relacionados con estos hallazgos histológicos y genéticos.
Estratificación del riesgo
Hay diferencias importantes en la estratificación del riesgo que los grupos de estudio de cáncer infantil usan para determinar el tratamiento, esto dificulta la comparación de los resultados de los distintos tratamientos administrados. En el Cuadro 5 se puede observar la variabilidad en las definiciones de los grupos de riesgo.
| COG (AHEP-0731) | SIOPEL (SIOPEL-3, 3HR, 4, 6) | GPOH | JPLT (JPLT-2 y 3) | |
|---|---|---|---|---|
| AFP = alfafetoproteína; COG = Children's Oncology Group; GPOH = Gesellschaft für Pädiatrische Onkologie und Hämatologie (Society for Paediatric Oncology and Haematology); JPLT = Japanese Study Group for Pediatric Liver Tumor; PRETEXT = PRE-Treatment EXTent of disease; SIOPEL = International Childhood Liver Tumors Strategy Group. | ||||
| aAdaptación de Czauderna et al. | ||||
| bPara obtener más información sobre las anotaciones utilizadas en PRETEXT, consultar el Cuadro 2. | ||||
| cLas definiciones del COG y PRETEXT sobre el compromiso vascular difieren. | ||||
| Riesgo muy bajo | PRETEXT I o II; subtipo histológico fetal bien diferenciado; resección primaria en el momento del diagnóstico | |||
| Riesgo bajo o riesgo estándar | PRETEXT I o II de cualquier subtipo histológico con resección primaria en el momento del diagnóstico | PRETEXT I, II o III | PRETEXT I, II, o III | PRETEXT I, II, o III |
| Riesgo intermediob | PRETEXT II, III o IV con tumor irresecable en el momento del diagnóstico; o V+c, P+, E+ | PRETEXT IV o cualquier PRETEXT con ruptura del tumor; o N1, P2, P2a, V3, V3a; o multifocal | ||
| Riesgo altob | Cualquier PRETEXT con M+; concentración de AFP <100 ng/ml | Cualquier PRETEXT; V+, P+, E+, M+; concentración de AFP <100 ng/ml; ruptura del tumor | Cualquier PRETEXT con V+, E+, P+, M+ o multifocal | Cualquier PRETEXT con M1 o N2; o concentración de AFP <100 ng/ml |
Modelo internacional de clasificación del riesgo
El grupo CHIC formuló un sistema novedoso de estratificación del riesgo para su uso en ensayos clínicos internacionales de acuerdo con las características pronósticas presentes en el momento del diagnóstico. El CHIC uniformó las diferentes definiciones y los sistemas de estadificación utilizados por los grupos de ensayos multicéntricos de cooperación del ámbito pediátrico, a fin de permitir la comparación de estudios dirigidos por grupos heterogéneos en distintos países. Se extrajeron los datos clínicos detallados originales de pacientes de 8 ensayos clínicos publicados donde se hizo revisión central de las imágenes y de los estudios histológicos, y se identificaron factores pronósticos mediante análisis univariante.
Según el análisis univariante inicial de los datos combinados con los patrones tradicionales de tratamiento clínico y los datos de ensayos clínicos grandes previos, se seleccionaron 5 grupos básicos, lo que permitió una estratificación del riesgo aún mayor. En el análisis multivariante posterior que se hizo de acuerdo a estos grupos básicos se definieron los siguientes factores clínicos pronósticos: Grupo PRETEXT (I, II, III o IV), presencia de metástasis (sí o no) y AFP (≤100 ng/ml). Los grupos principales son los siguientes:
- Grupo básico 1: PRETEXT I/II, no metastásico, AFP mayor de 100 ng/ml.
- Grupo básico 2: PRETEXT III, no metastásico, AFP mayor de 100 ng/ml.
- Grupo básico 3: PRETEXT IV, no metastásico, AFP mayor de 100 ng/ml.
- Grupo básico 4: Cualquier grupo PRETEXT, enfermedad metastásica en el momento del diagnóstico, AFP mayor de 100 ng/ml.
- Grupo básico 5: Cualquier grupo PRETEXT, metastásico o no metastásico, AFP de 100 ng/ml o menos en el momento del diagnóstico.
Se consultaron otros factores diagnósticos (por ejemplo, la edad) para cada una de las categorías principales, como la presencia de al menos una de las siguientes anotaciones PRETEXT (definidas como VPEFR+, ver Cuadro 2) o AFP menor o igual a 100 ng/ml:
- V: compromiso de la vena cava, compromiso de las 3 venas hepáticas, o ambos.
- P: compromiso de la bifurcación portal, compromiso de las venas portales derecha e izquierda, o ambos.
- E: diseminación tumoral extrahepática adyacente.
- F: tumor hepático multifocal.
- R: ruptura del tumor en el momento del diagnóstico.
Para los pacientes de los grupos PRETEXT I y II, se añadió una evaluación de la resecabilidad quirúrgica en el momento del diagnóstico. Los pacientes de cada una de las 5 categorías de los grupos básicos se estratificaron en subcategorías a partir de un análisis multivariante de eliminación gradual retrógrada usando otras características del paciente, como la edad y la presencia o ausencia de factores de anotación de PRETEXT (V, P, E, F y R). Cada una de estas subcategorías recibió 1 de 4 designaciones de riesgo (muy bajo, bajo, intermedio o alto). El resultado del análisis multivariante se utilizó para asignar a los pacientes a categorías de riesgo muy bajo, bajo, intermedio y alto, como se observa en la Figura 2. Por ejemplo, el hallazgo de una concentración de AFP de 100 ng/ml a 1000 ng/ml fue significativo solo en pacientes menores de 8 años en el grupo básico de PRETEXT III. El análisis permitirá asignar los grupos de riesgo con pronóstico similar a los grupos de tratamiento apropiados en los próximos protocolos internacionales.
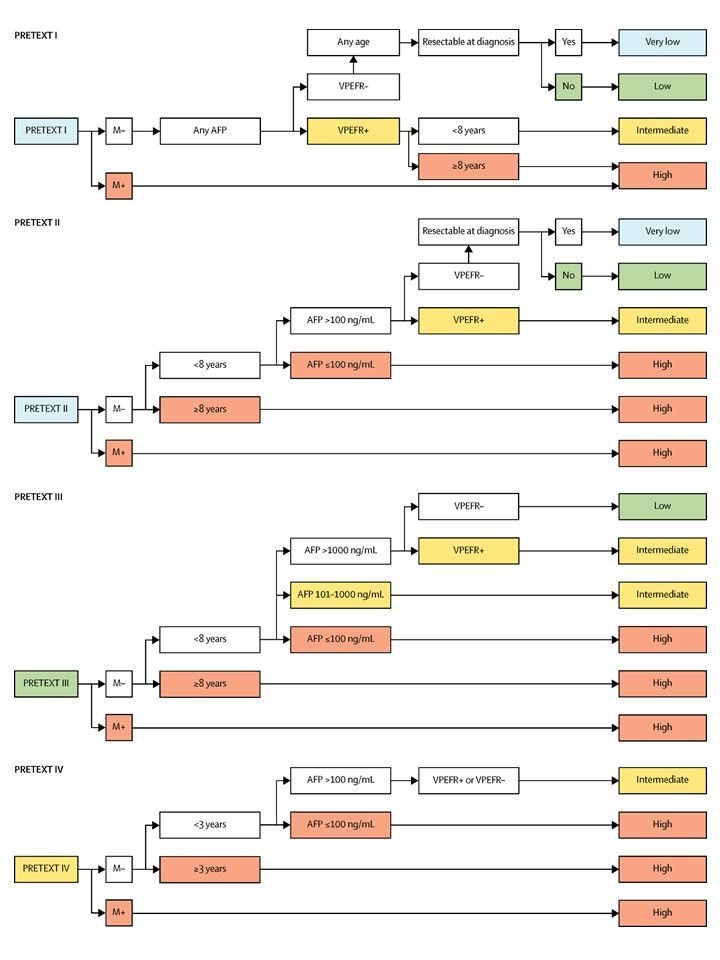 Figura 2. Árboles de estratificación del riesgo según la Children’s Hepatic tumors International Collaboration—Hepatoblastoma Stratification (CHIC-HS). Los grupos de riesgo muy bajo y riesgo bajo se separan solo por la resecabilidad en el momento del diagnóstico que se definió por consenso internacional como parte de las pautas quirúrgicas para el próximo ensayo colaborativo: Paediatric Hepatic International Tumour Trial (PHITT). Se emplean árboles de estratificación diferentes para cada uno de los cuatro grupos PRETEXT. AFP = alfafetoproteína. M = enfermedad metastásica. PRETEXT = PRETreatment EXTent of disease (alcance de la enfermedad antes del tratamiento). VPEFR: venoso, portal, extrahepático, multifocal y con ruptura; years: años; intermediate: intermedio; high: alto; any age: cualquier edad, resectable at diagnosis: resecable en el momento del diagnóstico; very low: muy bajo; low: bajo. Reproducción autorizada por Elsevier de The Lancet Oncology, Volume 18, Meyers RL, Maibach R, Hiyama E, Häberle B, Krailo M, Rangaswami A, Aronson DC, Malogolowkin MH, Perilongo G, von Schweinitz D, Ansari M, Lopez-Terrada D, Tanaka Y, Alaggio R, Leuschner I, Hishiki T, Schmid I, Watanabe K, Yoshimura K, Feng Y, Rinaldi E, Saraceno D, Derosa M, Czauderna P, Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children's Hepatic tumors International Collaboration, páginas 122–131, Derechos de autor (2017).
Figura 2. Árboles de estratificación del riesgo según la Children’s Hepatic tumors International Collaboration—Hepatoblastoma Stratification (CHIC-HS). Los grupos de riesgo muy bajo y riesgo bajo se separan solo por la resecabilidad en el momento del diagnóstico que se definió por consenso internacional como parte de las pautas quirúrgicas para el próximo ensayo colaborativo: Paediatric Hepatic International Tumour Trial (PHITT). Se emplean árboles de estratificación diferentes para cada uno de los cuatro grupos PRETEXT. AFP = alfafetoproteína. M = enfermedad metastásica. PRETEXT = PRETreatment EXTent of disease (alcance de la enfermedad antes del tratamiento). VPEFR: venoso, portal, extrahepático, multifocal y con ruptura; years: años; intermediate: intermedio; high: alto; any age: cualquier edad, resectable at diagnosis: resecable en el momento del diagnóstico; very low: muy bajo; low: bajo. Reproducción autorizada por Elsevier de The Lancet Oncology, Volume 18, Meyers RL, Maibach R, Hiyama E, Häberle B, Krailo M, Rangaswami A, Aronson DC, Malogolowkin MH, Perilongo G, von Schweinitz D, Ansari M, Lopez-Terrada D, Tanaka Y, Alaggio R, Leuschner I, Hishiki T, Schmid I, Watanabe K, Yoshimura K, Feng Y, Rinaldi E, Saraceno D, Derosa M, Czauderna P, Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children's Hepatic tumors International Collaboration, páginas 122–131, Derechos de autor (2017).
Tratamiento del hepatoblastoma
Las opciones de tratamiento del hepatoblastoma recién diagnosticado dependen de los siguientes aspectos:
- Si es posible extirpar el cáncer en el momento del diagnóstico.
- Subtipo histológico del tumor.
- Forma en que el cáncer responde a la quimioterapia.
- Si el cáncer metastatizó.
La quimioterapia a base de cisplatino produjo una tasa de supervivencia de más del 90 % en los niños con enfermedad resecable PRETEXT y POST-Treatment EXTent (POSTTEXT) I y II antes o después de la quimioterapia.
Los regímenes quimioterapéuticos que se usan para el tratamiento del hepatoblastoma y sus respectivos desenlaces se describen en el Cuadro 6 Para obtener información sobre cada estadio, consultar la sección Estratificación tumoral por imágenes y estadificación quirúrgica de Evans para el cáncer de hígado infantil.
| Estudio | Régimen quimioterapéutico | Número de pacientes | Resultados |
|---|---|---|---|
| AFP = alfafetoproteína; C5V = cisplatino, fluorouracilo (5-FU) y vincristina; CARBO = carboplatino; CCG = Children’s Cancer Group; CDDP = cisplatino; CITA = pirarrubicina y cisplatino; COG = Children's Oncology Group; DOXO = doxorrubicina; SSC = supervivencia sin complicaciones, GPOH = Gesellschaft für Pädiatrische Onkologie und Hämatologie (Society for Paediatric Oncology and Haematology); H+ = ruptura o hemorragia intraperitoneal; RA = riesgo alto; IFOS = ifosfamida; IPA = ifosfamida, cisplatino y doxorrubicina; ITEC = ifosfamida, pirarrubicina, etopósido, y carboplatino; JPLT = Japanese Study Group for Pediatric Liver Tumor; RB = riesgo bajo; SN = sin notificación; SG = supervivencia general; PLADO = cisplatino y doxorrubicina; POG = Pediatric Oncology Group; PRETEXT = PRE-Treatment EXTent of disease; SIOPEL = International Childhood Liver Tumors Strategy Group; RE = riesgo estándar; SUPERPLADO = cisplatino, doxorrubicina y carboplatino; THP = tetrahidropiranilo y Adriamycin (pirarrubicina); VP = vinorelbina y cisplatino; VPE+ = compromiso venoso, portal y extrahepático; VP16 = etopósido. | |||
| aAdaptación de Czauderna et al., Meyers et al., y Malogolowkin et al. | |||
| bEl estudio se cerró antes de tiempo debido a los desenlaces inferiores en el grupo de CDDP/CARBO. | |||
| INT0098 (CCG/POG) 1989–1992 | C5V vs. CDDP/DOXO | Estadios I/II: 50 | SSC/SG a 4 años: |
| I/II = 88 %/100 % vs. 96 %/96 % | |||
| Estadio III: 83 | III = 60 %/68 % vs. 68 %/71 % | ||
| Estadio IV: 40 | IV = 14 %/33 % vs. 37 %/42 % | ||
| P9645 (COG)b 1999–2002 | C5V vs. CDDP/CARBO | Estadio III: 38 | SSC/SG a 3 años: |
| III/IV: C5V = 60 %/74 %; CDDP/CARBO = 38 %/54 % | |||
| Estadio IV: 50 | |||
| AHEP0731 (COG) 2010–2014 [Nivel de evidencia C1] | RB: C5V (2 ciclos) | RB (estadios I/II): 49 | SSC a 5 años: 88 %; SG a 5 años: 91 % |
| HB 94 (GPOH); 1994–1997 | I/II: IFOS/CDDP/DOXO | Estadio I: 27 | SSC/SG a 4 años: |
| I = 89 %/96 % | |||
| Estadio II: 3 | II = 100 %/100 % | ||
| III/IV: IFOS/CDDP/DOXO + VP/CARBO | Estadio III: 25 | III = 68 %/76 % | |
| Estadio IV: 14 | IV = 21 %/36 % | ||
| HB 99 (GPOH); 1999–2004 | RE: IPA | RE: 58 | SSC/SG a 3 años: |
| RE = 90 %/88 % | |||
| RA: CARBO/VP16 | RA: 42 | CRI: = 52 %/55 % | |
| SIOPEL-2 1994–1998 | RE: PLADO | PRETEXT I: 6 | SSC/SG a 3años: |
| RE: 73 %/91 % | |||
| PRETEXT II: 36 | |||
| PRETEXT III: 25 | |||
| RA: CDDP/CARBO/DOXO | PRETEXT IV: 21 | RA: IV = 48 %/61 % | |
| Metástasis: 25 | RA: metástasis = 36 %/44 % | ||
| SIOPEL-3 1998–2006 | RE: CDDP vs. PLADO | RE: PRETEXT I: 18 | SSC/SG a 3 años: |
| RE: CDDP = 83 %/95 %; PLADO = 85 %/93 % | |||
| PRETEXT II: 133 | |||
| PRETEXT III: 104 | |||
| RA: SUPERPLADO | RA: PRETEXT IV: 74 | RA: General = 65 %/69 % | |
| VPE+: 70 | |||
| Metástasis. 70 | Metástasis = 57 %/63 % | ||
| AFP <100 ng/ml: 12 | |||
| SIOPEL-4 2005–2009 | RA: Bloque A: Semanal; CDDP/3 semanal DOXO; Bloque B: CARBO/DOXO | PRETEXT I: 2 | SSC/SG a 3 años: |
| Todos RA = 76 %/83 % | |||
| PRETEXT II: 17 | |||
| PRETEXT III: 27 | |||
| PRETEXT IV: 16 | RA: IV = 75 %/88 % | ||
| Metástasis: 39 | RA: Metástasis = 77 %/79 % | ||
| JPLT-1 1991–1999 | I/II: CDDP(30)/THP-DOXO | Estadio I: 9 | SSC/SG a 5 años: |
| I = SN/100 % | |||
| Estadio II: 32 | II = SN/76 % | ||
| III/IV: CDDP(60)/THP-DOXO | Estadio IIIa: 48 | IIIa = SN/50 % | |
| Estadio IIIb: 25 | IIIb = SN/64 % | ||
| Estadio IV: 20 | IV = SN/77 % | ||
| JPLT-2 1999–2010 [Nivel de evidencia C1] | Cirugía inicial y 2 ciclos de CITA | Estrato 1: PRETEXT I/II, sin factores de anotación excepto por H+ (n = 40) | SSC/SG a 5 años: |
| 74,2 %/89,9 % | |||
| 2 ciclos de CITA seguidos de cirugía y 2–4 ciclos de CITA | Estrato 2: PRETEXT II con multifocalidad (n = 80) | 84,8 %/90,8 % | |
| 2 ciclos de CITA seguidos de 2 ciclos de CITA (pacientes que responden al tratamiento); intento de cirugía que incluye trasplante | Estrato 3: PRETEXT I/II (con factores de anotación) y III/IV (n = 176) que responden al tratamiento | 71,6%/85,9% | |
| 2 ciclos de CITA seguidos de 2 ciclos de ITEC (que no responden al tratamiento); intento de cirugía que incluye trasplante | Estrato 4: PRETEXT I/II (con factores de anotación) y III/IV (n = 59) que no responden al tratamiento | 59,1 %/67,3 % | |
Opciones de tratamiento del hepatoblastoma resecable en el momento del diagnóstico
Cerca del 20 % al 30 % de los niños con hepatoblastoma presentan enfermedad resecable en el momento del diagnóstico. En el apéndice de las directrices quirúrgicas del Children's Oncology (COG) (AHEP0731 [NCT00980460]), se recomienda la resección del tumor en el momento del diagnóstico sin quimioterapia preoperatoria en niños con tumores del grupo PRETEXT I y tumores del grupo PRETEXT II con márgenes radiográficos de más de 1 cm de la vena cava y de las venas hepáticas media y porta. Los desenlaces para los pacientes después de someterse a una resección completa en el momento del diagnóstico en comparación con los pacientes con márgenes microscópicos positivos en el momento de la resección, son similares después de recibir quimioterapia.; [Nivel de evidencia C1]
Según el subtipo histológico, el pronóstico varía de la siguiente manera:
- Los pacientes con el subtipo histológico fetal bien diferenciado (4 % de los hepatoblastomas) tienen una tasa de SG a 3 y 5 años del 100 % con quimioterapia adyuvante mínima o sin esta.
- Los pacientes con hepatoblastomas de otro subtipo histológico diferente al fetal bien diferenciado o al indiferenciado de células pequeñas tienen una tasa de SG a 3 y 4 años de un 90 % a un 100 % con quimioterapia adyuvante.
- Si hay algún elemento indiferenciado de células pequeñas, la tasa de supervivencia a 3 años es del 40 % al 70 %.
Las opciones de tratamiento del hepatoblastoma resecable en el momento del diagnóstico y de subtipo diferente al fetal bien diferenciado son las siguientes:
- Resección seguida de 2 a 4 ciclos de quimioterapia.
Es posible que no sea necesaria una nueva resección de márgenes con compromiso microscópico. No se dispone de evidencia concluyente sobre los tumores resecados en el momento del diagnóstico en comparación con aquellos que presentan márgenes con compromiso microscópico resecados después de la quimioterapia preoperatoria.
Evidencia (resección quirúrgica macroscópica [con márgenes con compromiso microscópico o sin este] y quimioterapia posoperatoria):
- En el ensayo del COG AHEP0731 (NCT00980460), 49 de 51 pacientes con hepatoblastoma en estadio I o estadio II (sin características histológicas fetales puras) recibieron 2 ciclos de quimioterapia adyuvante con cisplatino, fluorouracilo y vincristina.[Nivel de evidencia C1]
- La tasa de SSC a 5 años fue del 88 % y la tasa de SG a 5 años fue del 91 %.
- Este desenlace es comparable a los desenlaces de niños tratados con 4 ciclos después de la resección inicial, así como a los desenlaces de los niños tratados con 2 ciclos de quimioterapia neoadyuvante antes de la resección seguida de 2 ciclos de quimioterapia después de la resección.
- No se dispone de datos confiables sobre el riesgo de recidiva local en pacientes con márgenes con compromiso microscópico que se resecaron en el momento del diagnóstico. Los estudios de SIOPEL indican que en los pacientes que recibieron quimioterapia preoperatoria, la presencia de algún margen con compromiso microscópico no aumentó el riesgo de recidiva local.; [Nivel de evidencia C1]
- En un estudio europeo realizado entre 1990 y 1994, 11 pacientes presentaron tejido tumoral en los márgenes quirúrgicos luego de una resección hepática y 2 pacientes murieron; ninguno presentó recidiva local. De los 11 pacientes, ninguno se sometió a una segunda resección y solo 1 paciente recibió radioterapia posoperatoria. Todos los pacientes recibieron 4 ciclos de cisplatino y doxorrubicina antes de la cirugía y 2 cursos de quimioterapia posoperatoria.
- En otro estudio europeo de hepatoblastoma de riesgo alto, 11 pacientes presentaron tumor residual microscópico después de la cirugía inicial y recibieron de 2 a 4 ciclos de quimioterapia posoperatoria sin cirugía adicional. De estos 11 pacientes, 9 sobrevivieron.
- En el estudio SIOPEL-2, sobrevivieron 13 de 13 pacientes con márgenes de resección con compromiso microscópico.
- En un estudio retrospectivo no planificado de los ensayos SIOPEL-2 y SIOPEL-3, se encontró que después de 4 cursos de cisplatino administrados a pacientes de riesgo estándar y 7 cursos de cisplatino alternado con doxorrubicina y carboplatino para los pacientes de riesgo alto, se realizó una resección cuando, a partir de las imágenes, se determinó que no sería peligrosa. De los 431 niños tratados en estos ensayos, 58 exhibieron márgenes tumorales con compromiso microscópico y 371 presentaron remisión completa. No hubo diferencias estadísticamente significativas en las tasas de recidiva local, SSC o SG entre los dos grupos.[Nivel de evidencia C1]
- En un ensayo clínico aleatorizado se demostró una eficacia comparable de la administración de cisplatino, vincristina y fluorouracilo posoperatorios y la administración de cisplatino y doxorrubicina para el tratamiento de pacientes con hepatoblastoma.
- Si bien los desenlaces de supervivencia fueron nominalmente más altos en los niños que recibieron cisplatino y doxorrubicina, esta diferencia no fue estadísticamente significativa.
- La administración de una combinación de cisplatino, vincristina y fluorouracilo presentó una toxicidad significativamente inferior a la administración de dosis de cisplatino y doxorrubicina.
Los resultados de los ensayos clínicos de quimioterapia se describen en el Cuadro 6.
Las opciones de tratamiento del hepatoblastoma de subtipo histológico fetal bien diferenciado (fetal puro) resecable en el momento del diagnóstico son las siguientes:
- Resección quirúrgica completa seguida de conducta expectante o quimioterapia.
Evidencia (resección quirúrgica completa seguida de conducta expectante o quimioterapia):
- En un ensayo clínico prospectivo del COG (INT0098), 9 niños con tumor en estadio I de subtipo histológico fetal bien diferenciado (completamente resecados) y menos de 2 mitosis por campo de gran aumento recibieron 4 ciclos de doxorrubicina adyuvante.
- Al cabo de una mediana de seguimiento de 5,1 años, las tasas de SSC y SG fueron del 100 % en los 9 niños.
- En el estudio del COG P9645 (NCT00003994), 16 pacientes con tumores en estadio I (completamente resecados) de tipo histológico fetal bien diferenciado no recibieron quimioterapia adyuvante. En 21 de estos 25 pacientes se estableció de manera retrospectiva y usando datos adecuados la clasificación PRETEXT y se encontraron tumores PRETEXT I, II y III en 7, 10 y 4 pacientes, respectivamente.
- Las tasas de SSC y SG fueron del 100 % en los pacientes con tumores del subtipo histológico fetal bien diferenciado en estadio I, incluso 1 paciente que se sometió a una segunda cirugía para tratar un margen tumoral comprometido.
Opciones de tratamiento del hepatoblastoma irresecable o que no se resecó en el momento del diagnóstico
Cerca del 70 % al 80 % de los niños con hepatoblastoma tienen tumores que no se resecaron en el momento del diagnóstico. En el apéndice AHEP0731 [NCT00980460] de las directrices quirúrgicas del COG, se recomienda una biopsia diagnóstica sin intentar resecar el tumor en niños con tumores PRETEXT II con menos de 1 cm de margen radiográfico en la vena cava y la vena hepática media, así como en todos los niños con tumores PRETEXT III y IV.
Las opciones de tratamiento del hepatoblastoma irresecable o que no se resecó en el momento del diagnóstico son las siguientes:
- Quimioterapia seguida de una nueva evaluación de la resecabilidad quirúrgica y resección quirúrgica completa.
- Quimioterapia seguida de una nueva evaluación de la resecabilidad quirúrgica y trasplante ortotópico de hígado.
- Quimioembolización transarterial (QETA) y radioembolización transarterial (RETA). La QETA y la RETA se pueden usar para mejorar la resecabilidad antes de la cirugía definitiva.
La ruptura tumoral en el momento de la presentación inicial, que produce una hemorragia importante que se puede controlar mediante embolización arterial transcatéter o resección parcial para estabilizar al paciente, no impide un desenlace favorable cuando se sigue de quimioterapia y cirugía definitiva.
En años recientes, la mayoría de los niños con hepatoblastoma se han tratado con quimioterapia. En los centros oncológicos europeos, los niños con hepatoblastoma resecable en el momento del diagnóstico reciben quimioterapia preoperatoria, lo que en ocasiones reduce la incidencia de complicaciones quirúrgicas en el momento de la resección. Se ha observado que el tratamiento con quimioterapia preoperatoria beneficia a los niños con hepatoblastoma. En contraste, en un estudio intergrupal estadounidense sobre el tratamiento de niños con hepatoblastomas, se alentó la resección en el momento del diagnóstico para todos los tumores susceptibles de resección sin riesgo excesivo. En el estudio (COG-P9645) los niños con tumores de subtipo histológico fetal bien diferenciado en estadio l no recibieron quimioterapia preoperatoria o posoperatoria, a menos que presentaran enfermedad progresiva. En este estudio, la mayoría de los pacientes con tumores PRETEXT III y todos los que tenían tumores PRETEXT IV recibieron quimioterapia antes de la resección o el trasplante.
Los pacientes cuyos tumores permanecen irresecables después de la quimioterapia se deben considerar candidatos para un trasplante de hígado. Cuando hay características que predicen irresecabilidad, es de suma importancia la pronta coordinación con un servicio de trasplante de hígado en pediatría. En el estudio del COG AHEP0731 (NCT00980460), se recomendó la derivación temprana (es decir, según las imágenes posteriores al segundo ciclo de quimioterapia) a un centro especializado en hígado con capacidad de realizar trasplantes para los pacientes con tumores POSTTEXT III con factor de anotación V o P y tumores POSTTEXT IV con factor de anotación F.
Evidencia (quimioterapia seguida de una nueva evaluación de la resecabilidad quirúrgica y resección quirúrgica completa o trasplante de hígado):
- En el estudio SIOPEL-1, la quimioterapia preoperatoria (doxorrubicina y cisplatino) se administró a todos los niños con hepatoblastoma con metástasis o sin esta. Después de la quimioterapia, y excluyendo a quienes se sometieron a un trasplante de hígado (<5 % de los pacientes), se realizó una resección completa.
- La quimioterapia se toleró bien.
- La resección completa se obtuvo en el 87 % de los niños.
- Esta estrategia produjo una SG del 75 % a los 5 años del diagnóstico.
- Se observaron resultados idénticos en un estudio internacional de seguimiento (SIOPEL-2).
- En el estudio SIOPEL-3, se comparó la administración de cisplatino solo con la administración de cisplatino y doxorrubicina en pacientes con hepatoblastoma de riesgo preoperatorio estándar. El riesgo estándar se definió como un tumor confinado al hígado y que no afectó más de 3 sectores.[Nivel de evidencia A1]
- Las tasas de resección y SG fueron similares en los grupos de cisplatino (95 %) y cisplatino con doxorrubicina (93 %).
- En un estudio piloto, SIOPEL-3HR, se administró cisplatino alternado con carboplatino y doxorrubicina en dosis intensivas a pacientes de riesgo alto con hepatoblastoma.
- De 74 pacientes con tumores PRETEXT IV (22 de los cuales también tenían metástasis), 31 pacientes tenían tumores que se volvieron resecables y 26 pacientes se sometieron a trasplante. La tasa del SG a 3 años para este grupo fue del 69 % (± 11 %).
- En los 70 pacientes con metástasis inscritos en este ensayo, la tasa de SSC a 3 años fue del 56 % y la tasa de SG fue del 62 %. De los pacientes con metástasis pulmonares, el 50 % logró la remisión completa de las metástasis con quimioterapia sola (sin cirugía pulmonar).
- El estudio SIOPEL-4 (NCT00077389) fue un ensayo multinacional de viabilidad de la administración de quimioterapia con dosis densas de cisplatino y doxorrubicina, y cirugía radical para un grupo de niños con hepatoblastoma de riesgo alto. En la medida de lo posible, se realizó la resección quirúrgica de todas las lesiones tumorales restantes después de la quimioterapia (incluso trasplante de hígado y metastasectomía, cuando fue necesario). Los pacientes cuyos tumores se resecaron o que se sometieron a un trasplante de hígado después de 3 ciclos de quimioterapia, más tarde recibieron 2 ciclos posoperatorios de carboplatino y doxorrubicina. Los pacientes cuyos tumores permanecieron irresecables después de 3 ciclos de quimioterapia recibieron 2 ciclos muy intensivos de carboplatino y doxorrubicina antes de la cirugía. Las masas tumorales primarias se clasificaron como PRETEXT II (27 %), III (44 %) y IV (26 %).[Nivel de evidencia B4]
- El 97 % (60 de 61) de los pacientes presentaron respuesta parcial a la quimioterapia.
- El 85 % (53) de los pacientes se sometieron a resección macroscópica completa; se encontró tumor microscópico en 5 pacientes, todos sobrevivientes sin enfermedad.
- De los pacientes, 2 murieron después de la cirugía.
- Se realizaron 37 hepatectomías parciales y 16 trasplantes de hígado.
- En el estudio participaron 62 pacientes de riesgo alto; el 74 % de los pacientes (62–84 %) se sometió a resección.
- La tasa de supervivencia sin enfermedad (SSE) a 3 años fue del 76 % (IC 95 %, 65–87 %).
- La tasa de SG a 3 años fue del 83 % (IC 95 %, 73–93 %).
- De los 16 pacientes clasificados como PRETEXT IV, 11 se reclasificaron a un grupo más bajo después de la quimioterapia (6 pacientes a PRETEXT III, 4 pacientes a PRETEXT II y 1 paciente a PRETEXT I). De los tumores, 12 se volvieron resecables; y de estos, 4 pacientes, se sometieron a hepatectomía parcial mientras que 8 pacientes se sometieron a trasplante de hígado. En los pacientes que presentaron enfermedad PRETEXT IV:
- La tasa de SG a 3 años fue del 73 % (IC 95 %, 51–96 %).
- La tasa de SG a 3 años fue del 80 % (IC 95 %, 60–100 %).
- En cerca del 75 % de los niños y adolescentes con hepatoblastoma irresecable al inicio, los tumores se vuelven resecables gracias a la administración de quimioterapia preoperatoria a base de cisplatino, y entre un 60 % y un 65 % de los pacientes sobrevivirán sin enfermedad.
En los Estados Unidos, los tumores irresecables se han tratado con quimioterapia antes de la resección o el trasplante. Según las imágenes radiográficas, la mayoría de los hepatoblastomas en estadios III y IV se vuelven resecables luego de 2 ciclos de quimioterapia. También se ha utilizado una combinación de ifosfamida, cisplatino y doxorrubicina seguida de resección posinducción para el tratamiento de la enfermedad en estadio avanzado. En algunos centros también se utilizó la resección amplia de tumores POSTTEXT III y IV seleccionados en lugar de un trasplante de hígado. Otras opciones, como la RETA y la QETA, se han utilizado para achicar la masa residual tumoral. Es posible que la RETA también facilite la resección quirúrgica por achicamiento del tumor cuando se añade a la quimioterapia.
El COG llevó a cabo un ensayo de fase III de un solo grupo (AHEP0731 [NCT00980460]) para pacientes con hepatoblastoma de riesgo intermedio. En el estudio se incluyeron 93 pacientes con enfermedad no metastásica irresecable y 9 pacientes con resección completa en el momento del diagnóstico. Todos los tumores eran de subtipo histológico indiferenciado de células pequeñas. Se evaluó la viabilidad y eficacia de la adición de doxorrubicina al tratamiento estándar (cisplatino, fluorouracilo y vincristina). En los 93 pacientes con enfermedad irresecable al inicio, la tasa de SSC a 5 años fue del 85 % (intervalo de confianza [IC] 95 %, 79–93 %) y la tasa de SG fue del 95 % (IC 95 %, 87–98 %).
La quimioterapia seguida de QETA y luego ultrasonidos focalizados de alta intensidad, mostró resultados prometedores en China para los pacientes con tumores PRETEXT III y IV, algunos de los cuales eran resecables, pero los pacientes no se sometieron a resección quirúrgica debido a la negativa de los progenitores.
Opciones de tratamiento del hepatoblastoma con metástasis en el momento del diagnóstico
Los desenlaces de los pacientes con hepatoblastoma metastásico en el momento del diagnóstico son precarios, pero es posible la supervivencia a largo plazo y la curación. Las tasas de supervivencia a 3 y 5 años oscilan entre el 20 % y el 79 %. Hasta la fecha, los mejores desenlaces para los niños con hepatoblastoma metastásico se derivaron del tratamiento con dosis densas de cisplatino y doxorrubicina, aunque también se observó toxicidad significativa (ensayo SIOPEL-4 [NCT00077389]).[Nivel de evidencia B4]
Las opciones de tratamiento del hepatoblastoma metastásico en el momento del diagnóstico son las siguientes:
- Quimioterapia seguida de una nueva evaluación de la resecabilidad quirúrgica.
- Si el tumor primario y la enfermedad extrahepática (por lo común, nódulos pulmonares) son resecables luego de la quimioterapia, se hace una resección quirúrgica seguida de más quimioterapia.
- Si la enfermedad metastásica extrahepática está en remisión completa luego de la quimioterapia o de la resección quirúrgica de un nódulo pulmonar, pero el tumor primario permanece irresecable, se hace un trasplante ortotópico de hígado.
- Si la enfermedad metastásica extrahepática no es resecable o el paciente no es candidato para un trasplante, es posible que se indique más quimioterapia adicional, QETA, RETA o radioterapia.
El régimen de quimioterapia combinada estándar en América del Norte es de 4 ciclos de cisplatino, vincristina y fluorouracilo o doxorrubicina y cisplatino, seguido de un intento de resección completa del tumor. Si el tumor se extirpa en su totalidad, se suelen administrar 2 cursos posoperatorios de la misma quimioterapia. Se notificaron los resultados de los estudios para diferentes regímenes de quimioterapia. Para obtener más información, consultar el Cuadro 6.
La quimioterapia de dosis altas con rescate de células madre no es más eficaz que la quimioterapia multifarmacológica estándar.
Evidencia (quimioterapia para el tratamiento de la enfermedad metastásica en el momento del diagnóstico seguida de cirugía):
- Un subconjunto de 39 pacientes que presentaban metástasis desde el inicio participaron en el ensayo SIOPEL-4 (NCT00077389), un ensayo multinacional de viabilidad de quimioterapia con dosis densas de cisplatino y doxorrubicina y cirugía radical para un grupo de niños con hepatoblastoma de riesgo alto. Los pacientes cuyos tumores se resecaron o que se sometieron a un trasplante de hígado después de 3 ciclos de quimioterapia, recibieron más tarde 2 ciclos posoperatorios de carboplatino y doxorrubicina. Los pacientes cuyos tumores eran irresecables después de 3 ciclos de quimioterapia recibieron 2 ciclos adicionales de carboplatino y doxorrubicina muy intensivos antes de la cirugía.[Nivel de evidencia B4]
- Después de 3 ciclos de quimioterapia, se obtuvo respuesta completa (solo de las metástasis) en 20 de 39 pacientes y respuesta parcial en 18 de 39 pacientes. De los pacientes que lograron una respuesta completa, 19 estaban vivos sin enfermedad 3 años después del diagnóstico.
- De los pacientes que lograron una respuesta parcial, 7 se sometieron a metastasectomía cerca del momento de la resección o el trasplante de hígado, con una tasa de SG del 100 %. Además, otros 7 pacientes que tenían nódulos pulmonares residuales pequeños se sometieron a resección sin metastasectomía; de ellos, 6 pacientes evolucionaron bien y 1 paciente presentó una recidiva.
- Con el tiempo, 2 pacientes que tenían metástasis iniciales, recidivaron.
- Se necesitó trasplante de hígado, en lugar de resección sola, para el tratamiento de 7 de los 39 pacientes que presentaban metástasis.
- Del subgrupo de 39 pacientes que presentaban metástasis desde el inicio, la tasa de SSE a 3 años fue del 77 % (IC 95 %, 63–90 %),y la tasa de SG fue del 79 % (IC 95 %, 66–92 %).
En los pacientes con tumores primarios resecados, cualquier remanente de metástasis pulmonar se debe extirpar mediante cirugía, de ser posible. Es posible facilitar la resección de las metástasis pulmonares mediante la localización con aguja por tomografía computarizada o la administración preoperatoria de verde de indocianina con fluorescencia intraoperatoria. En una revisión de pacientes tratados en un ensayo intergrupal de los Estados Unidos, se indicó que la resección de la metástasis se puede realizar en el momento de la resección del tumor primario.[Nivel de evidencia C1]
Si la enfermedad extrahepática está en remisión completa después de la quimioterapia y el tumor primario sigue siendo irresecable, es posible realizar un trasplante ortotópico de hígado.
Se observan discrepancias en lo desenlaces de pacientes con metástasis pulmonares en el momento del diagnóstico que se someten a trasplante ortotópico de hígado luego de la resolución completa de la enfermedad pulmonar como reacción a la quimioterapia pretrasplante. En algunos estudios se notificaron desenlaces favorables para estos pacientes, mientras que en otros se han observado tasas altas de recidiva del hepatoblastoma. Todos estos estudios se ven limitados por el número bajo de pacientes. Se requieren más estudios para definir mejor los desenlaces de este subgrupo de pacientes. En ensayos clínicos recientes se produjeron pocas recidivas pulmonares en niños que presentaron enfermedad metastásica y se sometieron a trasplantes de hígado.
Si la enfermedad extrahepática no es resecable después de la quimioterapia o el paciente no es apto para un trasplante, los abordajes alternativos de tratamiento son los siguientes:
- Otros fármacos quimioterapéuticos. Se han utilizado fármacos quimioterapéuticos como irinotecán, dosis altas de cisplatino y etopósido o infusión continua de doxorrubicina.; [Nivel de evidencia C1]
- QETA.
- Radioterapia.
Tratamiento del hepatoblastoma progresivo o recidivante
El pronóstico de un paciente con hepatoblastoma progresivo o recidivante depende de varios factores, como los siguientes:
- Sitio de la recidiva.
- Tratamiento previo.
- Consideraciones individuales del paciente.
Las opciones de tratamiento del hepatoblastoma recidivante o progresivo son las siguientes:
- Resección quirúrgica. En los pacientes con hepatoblastoma que se resecó por completo en el momento del diagnóstico inicial, es posible que el tratamiento quirúrgico radical de las metástasis pulmonares aisladas que se presentan durante el curso de la enfermedad, junto con una estrategia general que incluya quimioterapia, haga posible una SSE prolongada.
De ser posible, las metástasis aisladas se deben resecar en su totalidad en aquellos pacientes cuyo tumor primario está controlado. En un estudio retrospectivo de pacientes de los estudios SIOPEL 1, 2 y 3, se observó una incidencia de recidiva del 12 % después de la remisión completa mediante imágenes y concentraciones de AFP. Los desenlaces después de la recidiva fueron mejores si el tumor era susceptible de cirugía. De los pacientes sometidos a quimioterapia y cirugía, la tasa de SSC a 3 años fue del 34 % y la tasa de SG fue del 43 %.[Nivel de evidencia C1]
Se debe considerar la participación en un ensayo clínico si no se puede extirpar toda la enfermedad recidivante mediante cirugía. Es posible que los ensayos clínicos de fase l y fase II sean apropiados.
- Quimioterapia. En los análisis de supervivencia después de la recidiva, se demostró que algunos pacientes tratados con cisplatino, vincristina y fluorouracilo podían recuperarse con regímenes que contenían doxorrubicina, pero los pacientes tratados con doxorrubicina y cisplatino no se podían recuperar con vincristina y fluorouracilo. La adición de doxorrubicina a la combinación de vincristina, fluorouracilo y cisplatino se evaluó clínicamente en el estudio del COG AHEP0731 (NCT00980460).
La combinación de vincristina e irinotecán y la administración de irinotecán en monoterapia se han utilizado con cierto éxito.; [Nivel de evidencia C1]
En una revisión de los estudios de fase I y II del COG, no se encontraron fármacos prometedores para el hepatoblastoma en recaída.
- Trasplante de hígado. Se debe considerar el trasplante de hígado para los pacientes con recidiva de enfermedad no metastásica en el hígado que no es susceptible de resección.
- Ablación percutánea. La ablación percutánea por radiofrecuencia se ha utilizado como alternativa a la resección quirúrgica del hepatoblastoma oligometastático.[Nivel de evidencia C1] También se pueden considerar técnicas de ablación percutánea como terapia paliativa.
Opciones de tratamiento en evaluación clínica para el hepatoblastoma
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de ensayo clínico nacional o institucional en curso:
- AHEP1531 (NCT03533582) (Cisplatin and Combination Chemotherapy in Treating Children and Young Adults with Hepatoblastoma or Liver Cancer After Surgery):
este ensayo representa la participación del COG en un gran ensayo internacional (PHITT) de tratamiento para todos los estadios de hepatoblastoma y carcinoma hepatocelular en niños.
- El hepatoblastoma de riesgo muy bajo se define como cualquiera de los siguientes casos: 1) masa de subtipo histológico fetal bien diferenciado resecada por completo en el momento del diagnóstico y pacientes que no reciben quimioterapia; 2) masa de subtipo histológico diferente al fetal bien diferenciado o masa de subtipo histológico fetal bien diferenciado pero con resección incompleta, y pacientes que reciben 2 ciclos de quimioterapia con cisplatino.
- El hepatoblastoma de riesgo bajo se define como un tumor PRETEXT I a III sin factores de anotación tipo VPEFR (compromiso venoso, compromiso portal, diseminación extrahepática, multifocalidad y ruptura tumoral). Estos pacientes se tratan con 2 ciclos de quimioterapia con cisplatino y luego se someten a resección (si es posible), seguida de asignación al azar a tratamientos de 2 o 4 ciclos adicionales de cisplatino. Si el tumor es irresecable, el paciente recibe 2 ciclos de quimioterapia con cisplatino adicionales y se hace una nueva evaluación de la resecabilidad del tumor. Si el tumor todavía es irresecable, los pacientes se someten a trasplante de hígado.
- El hepatoblastoma de riesgo intermedio se define como un tumor primario PRETEXT I a III con un factor de anotación VPEFR, pero sin metástasis. Los pacientes se asignan al azar a 4 ciclos de cisplatino de 14 días o 4 ciclos de 21 días de C5VD (cisplatino, fluorouracilo, vincristina y doxorrubicina). Los equipos de trasplante se consultan pronto según la necesidad. Luego, los pacientes de ambos grupos se someten a resección y reciben 2 ciclos adicionales de la quimioterapia asignada.
- El hepatoblastoma de riesgo alto se define por la presencia de metástasis a distancia, AFP menor de 100, u 8 años o más de edad. Todos los pacientes se tratan con 3 ciclos de la quimioterapia de inducción del ensayo SIOPEL-4 (dosis intensivas de cisplatino, doxorrubicina y carboplatino). Los pacientes de 8 años o más, aquellos con una AFP menor de 100 y los pacientes cuyas metástasis desaparecieron reciben 3 ciclos de carboplatino y doxorrubicina. Los pacientes con metástasis persistentes al final de la inducción se asignan al azar a recibir 6 ciclos de carboplatino y doxorrubicina alternados con carboplatino y etopósido, o 6 ciclos de carboplatino y doxorrubicina alternados con vincristina e irinotecán.
References
- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations). National Cancer Institute, 2012, Section 29. Also available online. Last accessed August 11, 2022.
- Bulterys M, Goodman MT, Smith MA, et al.: Hepatic tumors. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 91-98. Also available online. Last accessed August 11, 2022.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 15, 2023.
- Turcotte LM, Georgieff MK, Ross JA, et al.: Neonatal medical exposures and characteristics of low birth weight hepatoblastoma cases: a report from the Children's Oncology Group. Pediatr Blood Cancer 61 (11): 2018-23, 2014.
- Tanimura M, Matsui I, Abe J, et al.: Increased risk of hepatoblastoma among immature children with a lower birth weight. Cancer Res 58 (14): 3032-5, 1998.
- McLaughlin CC, Baptiste MS, Schymura MJ, et al.: Maternal and infant birth characteristics and hepatoblastoma. Am J Epidemiol 163 (9): 818-28, 2006.
- Darbari A, Sabin KM, Shapiro CN, et al.: Epidemiology of primary hepatic malignancies in U.S. children. Hepatology 38 (3): 560-6, 2003.
- Kamien BA, Gabbett MT: Aicardi syndrome associated with hepatoblastoma and pulmonary sequestration. Am J Med Genet A 149A (8): 1850-2, 2009.
- DeBaun MR, Tucker MA: Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr 132 (3 Pt 1): 398-400, 1998.
- Weksberg R, Shuman C, Smith AC: Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 137C (1): 12-23, 2005.
- Iwama T, Mishima Y: Mortality in young first-degree relatives of patients with familial adenomatous polyposis. Cancer 73 (8): 2065-8, 1994.
- Garber JE, Li FP, Kingston JE, et al.: Hepatoblastoma and familial adenomatous polyposis. J Natl Cancer Inst 80 (20): 1626-8, 1988.
- Giardiello FM, Petersen GM, Brensinger JD, et al.: Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut 39 (96): 867-9, 1996.
- Ito E, Sato Y, Kawauchi K, et al.: Type 1a glycogen storage disease with hepatoblastoma in siblings. Cancer 59 (10): 1776-80, 1987.
- Ikeda H, Hachitanda Y, Tanimura M, et al.: Development of unfavorable hepatoblastoma in children of very low birth weight: results of a surgical and pathologic review. Cancer 82 (9): 1789-96, 1998.
- Spector LG, Birch J: The epidemiology of hepatoblastoma. Pediatr Blood Cancer 59 (5): 776-9, 2012.
- Buonuomo PS, Ruggiero A, Vasta I, et al.: Second case of hepatoblastoma in a young patient with Simpson-Golabi-Behmel syndrome. Pediatr Hematol Oncol 22 (7): 623-8, 2005 Oct-Nov.
- Tan ZH, Lai A, Chen CK, et al.: Association of trisomy 18 with hepatoblastoma and its implications. Eur J Pediatr 173 (12): 1595-8, 2014.
- Trobaugh-Lotrario AD, Venkatramani R, Feusner JH: Hepatoblastoma in children with Beckwith-Wiedemann syndrome: does it warrant different treatment? J Pediatr Hematol Oncol 36 (5): 369-73, 2014.
- Hoyme HE, Seaver LH, Jones KL, et al.: Isolated hemihyperplasia (hemihypertrophy): report of a prospective multicenter study of the incidence of neoplasia and review. Am J Med Genet 79 (4): 274-8, 1998.
- Clericuzio CL, Martin RA: Diagnostic criteria and tumor screening for individuals with isolated hemihyperplasia. Genet Med 11 (3): 220-2, 2009.
- Algar EM, St Heaps L, Darmanian A, et al.: Paternally inherited submicroscopic duplication at 11p15.5 implicates insulin-like growth factor II in overgrowth and Wilms' tumorigenesis. Cancer Res 67 (5): 2360-5, 2007.
- Steenman M, Westerveld A, Mannens M: Genetics of Beckwith-Wiedemann syndrome-associated tumors: common genetic pathways. Genes Chromosomes Cancer 28 (1): 1-13, 2000.
- Albrecht S, Hartmann W, Houshdaran F, et al.: Allelic loss but absence of mutations in the polyspecific transporter gene BWR1A on 11p15.5 in hepatoblastoma. Int J Cancer 111 (4): 627-32, 2004.
- Clericuzio CL, Chen E, McNeil DE, et al.: Serum alpha-fetoprotein screening for hepatoblastoma in children with Beckwith-Wiedemann syndrome or isolated hemihyperplasia. J Pediatr 143 (2): 270-2, 2003.
- Mussa A, Molinatto C, Baldassarre G, et al.: Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J Pediatr 176: 142-149.e1, 2016.
- Aretz S, Koch A, Uhlhaas S, et al.: Should children at risk for familial adenomatous polyposis be screened for hepatoblastoma and children with apparently sporadic hepatoblastoma be screened for APC germline mutations? Pediatr Blood Cancer 47 (6): 811-8, 2006.
- Hirschman BA, Pollock BH, Tomlinson GE: The spectrum of APC mutations in children with hepatoblastoma from familial adenomatous polyposis kindreds. J Pediatr 147 (2): 263-6, 2005.
- Koch A, Denkhaus D, Albrecht S, et al.: Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res 59 (2): 269-73, 1999.
- Kalish JM, Doros L, Helman LJ, et al.: Surveillance Recommendations for Children with Overgrowth Syndromes and Predisposition to Wilms Tumors and Hepatoblastoma. Clin Cancer Res 23 (13): e115-e122, 2017.
- Eichenmüller M, Trippel F, Kreuder M, et al.: The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J Hepatol 61 (6): 1312-20, 2014.
- Jia D, Dong R, Jing Y, et al.: Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology 60 (5): 1686-96, 2014.
- Sumazin P, Chen Y, Treviño LR, et al.: Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology 65 (1): 104-121, 2017.
- Carrillo-Reixach J, Torrens L, Simon-Coma M, et al.: Epigenetic footprint enables molecular risk stratification of hepatoblastoma with clinical implications. J Hepatol 73 (2): 328-341, 2020.
- Gröbner SN, Worst BC, Weischenfeldt J, et al.: The landscape of genomic alterations across childhood cancers. Nature 555 (7696): 321-327, 2018.
- Sekiguchi M, Seki M, Kawai T, et al.: Integrated multiomics analysis of hepatoblastoma unravels its heterogeneity and provides novel druggable targets. NPJ Precis Oncol 4: 20, 2020.
- Cairo S, Armengol C, Maibach R, et al.: A combined clinical and biological risk classification improves prediction of outcome in hepatoblastoma patients. Eur J Cancer 141: 30-39, 2020.
- Cancer Genome Atlas Research Network. Electronic address: [email protected], Cancer Genome Atlas Research Network: Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 169 (7): 1327-1341.e23, 2017.
- Albrecht S, von Schweinitz D, Waha A, et al.: Loss of maternal alleles on chromosome arm 11p in hepatoblastoma. Cancer Res 54 (19): 5041-4, 1994.
- Cairo S, Armengol C, De Reyniès A, et al.: Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 14 (6): 471-84, 2008.
- Yoon JM, Burns RC, Malogolowkin MH, et al.: Treatment of infantile choriocarcinoma of the liver. Pediatr Blood Cancer 49 (1): 99-102, 2007.
- Boman F, Bossard C, Fabre M, et al.: Mesenchymal hamartomas of the liver may be associated with increased serum alpha foetoprotein concentrations and mimic hepatoblastomas. Eur J Pediatr Surg 14 (1): 63-6, 2004.
- Blohm ME, Vesterling-Hörner D, Calaminus G, et al.: Alpha 1-fetoprotein (AFP) reference values in infants up to 2 years of age. Pediatr Hematol Oncol 15 (2): 135-42, 1998 Mar-Apr.
- Wu JT, Book L, Sudar K: Serum alpha fetoprotein (AFP) levels in normal infants. Pediatr Res 15 (1): 50-2, 1981.
- Schneider DT, Calaminus G, Göbel U: Diagnostic value of alpha 1-fetoprotein and beta-human chorionic gonadotropin in infancy and childhood. Pediatr Hematol Oncol 18 (1): 11-26, 2001 Jan-Feb.
- Nakagawara A, Ikeda K, Tsuneyoshi M, et al.: Hepatoblastoma producing both alpha-fetoprotein and human chorionic gonadotropin. Clinicopathologic analysis of four cases and a review of the literature. Cancer 56 (7): 1636-42, 1985.
- Perilongo G, Malogolowkin M, Feusner J: Hepatoblastoma clinical research: lessons learned and future challenges. Pediatr Blood Cancer 59 (5): 818-21, 2012.
- Trobaugh-Lotrario AD, Katzenstein HM: Chemotherapeutic approaches for newly diagnosed hepatoblastoma: past, present, and future strategies. Pediatr Blood Cancer 59 (5): 809-12, 2012.
- Trobaugh-Lotrario AD, Chaiyachati BH, Meyers RL, et al.: Outcomes for patients with congenital hepatoblastoma. Pediatr Blood Cancer 60 (11): 1817-25, 2013.
- Czauderna P, Haeberle B, Hiyama E, et al.: The Children's Hepatic tumors International Collaboration (CHIC): Novel global rare tumor database yields new prognostic factors in hepatoblastoma and becomes a research model. Eur J Cancer 52: 92-101, 2016.
- Otte JB, Pritchard J, Aronson DC, et al.: Liver transplantation for hepatoblastoma: results from the International Society of Pediatric Oncology (SIOP) study SIOPEL-1 and review of the world experience. Pediatr Blood Cancer 42 (1): 74-83, 2004.
- Meyers RL, Rowland JR, Krailo M, et al.: Predictive power of pretreatment prognostic factors in children with hepatoblastoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 53 (6): 1016-22, 2009.
- Ortega JA, Douglass EC, Feusner JH, et al.: Randomized comparison of cisplatin/vincristine/fluorouracil and cisplatin/continuous infusion doxorubicin for treatment of pediatric hepatoblastoma: A report from the Children's Cancer Group and the Pediatric Oncology Group. J Clin Oncol 18 (14): 2665-75, 2000.
- Malogolowkin MH, Katzenstein HM, Meyers RL, et al.: Complete surgical resection is curative for children with hepatoblastoma with pure fetal histology: a report from the Children's Oncology Group. J Clin Oncol 29 (24): 3301-6, 2011.
- De Ioris M, Brugieres L, Zimmermann A, et al.: Hepatoblastoma with a low serum alpha-fetoprotein level at diagnosis: the SIOPEL group experience. Eur J Cancer 44 (4): 545-50, 2008.
- Pritchard J, Brown J, Shafford E, et al.: Cisplatin, doxorubicin, and delayed surgery for childhood hepatoblastoma: a successful approach--results of the first prospective study of the International Society of Pediatric Oncology. J Clin Oncol 18 (22): 3819-28, 2000.
- Perilongo G, Brown J, Shafford E, et al.: Hepatoblastoma presenting with lung metastases: treatment results of the first cooperative, prospective study of the International Society of Paediatric Oncology on childhood liver tumors. Cancer 89 (8): 1845-53, 2000.
- Perilongo G, Shafford E, Maibach R, et al.: Risk-adapted treatment for childhood hepatoblastoma. final report of the second study of the International Society of Paediatric Oncology--SIOPEL 2. Eur J Cancer 40 (3): 411-21, 2004.
- Zsíros J, Maibach R, Shafford E, et al.: Successful treatment of childhood high-risk hepatoblastoma with dose-intensive multiagent chemotherapy and surgery: final results of the SIOPEL-3HR study. J Clin Oncol 28 (15): 2584-90, 2010.
- Katzenstein HM, Furman WL, Malogolowkin MH, et al.: Upfront window vincristine/irinotecan treatment of high-risk hepatoblastoma: A report from the Children's Oncology Group AHEP0731 study committee. Cancer 123 (12): 2360-2367, 2017.
- O'Neill AF, Towbin AJ, Krailo MD, et al.: Characterization of Pulmonary Metastases in Children With Hepatoblastoma Treated on Children's Oncology Group Protocol AHEP0731 (The Treatment of Children With All Stages of Hepatoblastoma): A Report From the Children's Oncology Group. J Clin Oncol 35 (30): 3465-3473, 2017.
- Fuchs J, Rydzynski J, Von Schweinitz D, et al.: Pretreatment prognostic factors and treatment results in children with hepatoblastoma: a report from the German Cooperative Pediatric Liver Tumor Study HB 94. Cancer 95 (1): 172-82, 2002.
- Aronson DC, Schnater JM, Staalman CR, et al.: Predictive value of the pretreatment extent of disease system in hepatoblastoma: results from the International Society of Pediatric Oncology Liver Tumor Study Group SIOPEL-1 study. J Clin Oncol 23 (6): 1245-52, 2005.
- Meyers RL, Maibach R, Hiyama E, et al.: Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children's Hepatic tumors International Collaboration. Lancet Oncol 18 (1): 122-131, 2017.
- Haeberle B, Rangaswami A, Krailo M, et al.: The importance of age as prognostic factor for the outcome of patients with hepatoblastoma: Analysis from the Children's Hepatic tumors International Collaboration (CHIC) database. Pediatr Blood Cancer 67 (8): e28350, 2020.
- Dall'Igna P, Brugieres L, Christin AS, et al.: Hepatoblastoma in children aged less than six months at diagnosis: A report from the SIOPEL group. Pediatr Blood Cancer 65 (1): , 2018.
- Ortega JA, Krailo MD, Haas JE, et al.: Effective treatment of unresectable or metastatic hepatoblastoma with cisplatin and continuous infusion doxorubicin chemotherapy: a report from the Childrens Cancer Study Group. J Clin Oncol 9 (12): 2167-76, 1991.
- Douglass EC, Reynolds M, Finegold M, et al.: Cisplatin, vincristine, and fluorouracil therapy for hepatoblastoma: a Pediatric Oncology Group study. J Clin Oncol 11 (1): 96-9, 1993.
- Meyers RL, Czauderna P, Otte JB: Surgical treatment of hepatoblastoma. Pediatr Blood Cancer 59 (5): 800-8, 2012.
- Hiyama E, Hishiki T, Watanabe K, et al.: Resectability and tumor response after preoperative chemotherapy in hepatoblastoma treated by the Japanese Study Group for Pediatric Liver Tumor (JPLT)-2 protocol. J Pediatr Surg 51 (12): 2053-2057, 2016.
- Becker K, Furch C, Schmid I, et al.: Impact of postoperative complications on overall survival of patients with hepatoblastoma. Pediatr Blood Cancer 62 (1): 24-8, 2015.
- McAteer JP, Goldin AB, Healey PJ, et al.: Surgical treatment of primary liver tumors in children: outcomes analysis of resection and transplantation in the SEER database. Pediatr Transplant 17 (8): 744-50, 2013.
- Schnater JM, Aronson DC, Plaschkes J, et al.: Surgical view of the treatment of patients with hepatoblastoma: results from the first prospective trial of the International Society of Pediatric Oncology Liver Tumor Study Group. Cancer 94 (4): 1111-20, 2002.
- Zsiros J, Brugieres L, Brock P, et al.: Dose-dense cisplatin-based chemotherapy and surgery for children with high-risk hepatoblastoma (SIOPEL-4): a prospective, single-arm, feasibility study. Lancet Oncol 14 (9): 834-42, 2013.
- Van Tornout JM, Buckley JD, Quinn JJ, et al.: Timing and magnitude of decline in alpha-fetoprotein levels in treated children with unresectable or metastatic hepatoblastoma are predictors of outcome: a report from the Children's Cancer Group. J Clin Oncol 15 (3): 1190-7, 1997.
- Nguyen R, McCarville MB, Sykes A, et al.: Rapid decrease of serum alpha-fetoprotein and tumor volume predicts outcome in children with hepatoblastoma treated with neoadjuvant chemotherapy. Int J Clin Oncol 23 (5): 900-907, 2018.
- Brown J, Perilongo G, Shafford E, et al.: Pretreatment prognostic factors for children with hepatoblastoma-- results from the International Society of Paediatric Oncology (SIOP) study SIOPEL 1. Eur J Cancer 36 (11): 1418-25, 2000.
- D'Antiga L, Vallortigara F, Cillo U, et al.: Features predicting unresectability in hepatoblastoma. Cancer 110 (5): 1050-8, 2007.
- Czauderna P, Lopez-Terrada D, Hiyama E, et al.: Hepatoblastoma state of the art: pathology, genetics, risk stratification, and chemotherapy. Curr Opin Pediatr 26 (1): 19-28, 2014.
- López-Terrada D, Alaggio R, de Dávila MT, et al.: Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol 27 (3): 472-91, 2014.
- Weinberg AG, Finegold MJ: Primary hepatic tumors of childhood. Hum Pathol 14 (6): 512-37, 1983.
- Haas JE, Muczynski KA, Krailo M, et al.: Histopathology and prognosis in childhood hepatoblastoma and hepatocarcinoma. Cancer 64 (5): 1082-95, 1989.
- Rowland JM: Hepatoblastoma: assessment of criteria for histologic classification. Med Pediatr Oncol 39 (5): 478-83, 2002.
- Trobaugh-Lotrario AD, Tomlinson GE, Finegold MJ, et al.: Small cell undifferentiated variant of hepatoblastoma: adverse clinical and molecular features similar to rhabdoid tumors. Pediatr Blood Cancer 52 (3): 328-34, 2009.
- Gunawan B, Schäfer KL, Sattler B, et al.: Undifferentiated small cell hepatoblastoma with a chromosomal translocation t(22;22)(q11;q13). Histopathology 40 (5): 485-7, 2002.
- Trobaugh-Lotrario A, Katzenstein HM, Ranganathan S, et al.: Small Cell Undifferentiated Histology Does Not Adversely Affect Outcome in Hepatoblastoma: A Report From the Children's Oncology Group (COG) AHEP0731 Study Committee. J Clin Oncol 40 (5): 459-467, 2022.
- Conran RM, Hitchcock CL, Waclawiw MA, et al.: Hepatoblastoma: the prognostic significance of histologic type. Pediatr Pathol 12 (2): 167-83, 1992 Mar-Apr.
- Zhou S, Gomulia E, Mascarenhas L, et al.: Is INI1-retained small cell undifferentiated histology in hepatoblastoma unfavorable? Hum Pathol 46 (4): 620-4, 2015.
- Vokuhl C, Oyen F, Häberle B, et al.: Small cell undifferentiated (SCUD) hepatoblastomas: All malignant rhabdoid tumors? Genes Chromosomes Cancer 55 (12): 925-931, 2016.
- Cornet M, De Lambert G, Pariente D, et al.: Rhabdoid tumor of the liver: Report of 6 pediatric cases treated at a single institute. J Pediatr Surg 53 (3): 567-571, 2018.
- Meyers RL, Tiao G, de Ville de Goyet J, et al.: Hepatoblastoma state of the art: pre-treatment extent of disease, surgical resection guidelines and the role of liver transplantation. Curr Opin Pediatr 26 (1): 29-36, 2014.
- Malogolowkin MH, Katzenstein H, Krailo MD, et al.: Intensified platinum therapy is an ineffective strategy for improving outcome in pediatric patients with advanced hepatoblastoma. J Clin Oncol 24 (18): 2879-84, 2006.
- Katzenstein HM, Langham MR, Malogolowkin MH, et al.: Minimal adjuvant chemotherapy for children with hepatoblastoma resected at diagnosis (AHEP0731): a Children's Oncology Group, multicentre, phase 3 trial. Lancet Oncol 20 (5): 719-727, 2019.
- Hiyama E, Hishiki T, Watanabe K, et al.: Outcome and Late Complications of Hepatoblastomas Treated Using the Japanese Study Group for Pediatric Liver Tumor 2 Protocol. J Clin Oncol 38 (22): 2488-2498, 2020.
- Aronson DC, Weeda VB, Maibach R, et al.: Microscopically positive resection margin after hepatoblastoma resection: what is the impact on prognosis? A Childhood Liver Tumours Strategy Group (SIOPEL) report. Eur J Cancer 106: 126-132, 2019.
- Vasudevan SA, Meyers RL, Finegold MJ, et al.: Outcomes of children with well-differentiated fetal hepatoblastoma treated with surgery only: Report from Children's Oncology Group Trial, AHEP0731. J Pediatr Surg 57 (10): 251-256, 2022.
- Perilongo G, Maibach R, Shafford E, et al.: Cisplatin versus cisplatin plus doxorubicin for standard-risk hepatoblastoma. N Engl J Med 361 (17): 1662-70, 2009.
- Reyes JD, Carr B, Dvorchik I, et al.: Liver transplantation and chemotherapy for hepatoblastoma and hepatocellular cancer in childhood and adolescence. J Pediatr 136 (6): 795-804, 2000.
- Molmenti EP, Wilkinson K, Molmenti H, et al.: Treatment of unresectable hepatoblastoma with liver transplantation in the pediatric population. Am J Transplant 2 (6): 535-8, 2002.
- Czauderna P, Otte JB, Aronson DC, et al.: Guidelines for surgical treatment of hepatoblastoma in the modern era--recommendations from the Childhood Liver Tumour Strategy Group of the International Society of Paediatric Oncology (SIOPEL). Eur J Cancer 41 (7): 1031-6, 2005.
- Austin MT, Leys CM, Feurer ID, et al.: Liver transplantation for childhood hepatic malignancy: a review of the United Network for Organ Sharing (UNOS) database. J Pediatr Surg 41 (1): 182-6, 2006.
- Pham TA, Gallo AM, Concepcion W, et al.: Effect of Liver Transplant on Long-term Disease-Free Survival in Children With Hepatoblastoma and Hepatocellular Cancer. JAMA Surg 150 (12): 1150-8, 2015.
- Khan AS, Brecklin B, Vachharajani N, et al.: Liver Transplantation for Malignant Primary Pediatric Hepatic Tumors. J Am Coll Surg 225 (1): 103-113, 2017.
- Xianliang H, Jianhong L, Xuewu J, et al.: Cure of hepatoblastoma with transcatheter arterial chemoembolization. J Pediatr Hematol Oncol 26 (1): 60-3, 2004.
- Malogolowkin MH, Stanley P, Steele DA, et al.: Feasibility and toxicity of chemoembolization for children with liver tumors. J Clin Oncol 18 (6): 1279-84, 2000.
- Aguado A, Dunn SP, Averill LW, et al.: Successful use of transarterial radioembolization with yttrium-90 (TARE-Y90) in two children with hepatoblastoma. Pediatr Blood Cancer 67 (9): e28421, 2020.
- Madanur MA, Battula N, Davenport M, et al.: Staged resection for a ruptured hepatoblastoma: a 6-year follow-up. Pediatr Surg Int 23 (6): 609-11, 2007.
- Aronson DC, Meyers RL: Malignant tumors of the liver in children. Semin Pediatr Surg 25 (5): 265-275, 2016.
- Venkatramani R, Stein JE, Sapra A, et al.: Effect of neoadjuvant chemotherapy on resectability of stage III and IV hepatoblastoma. Br J Surg 102 (1): 108-13, 2015.
- von Schweinitz D, Hecker H, Harms D, et al.: Complete resection before development of drug resistance is essential for survival from advanced hepatoblastoma--a report from the German Cooperative Pediatric Liver Tumor Study HB-89. J Pediatr Surg 30 (6): 845-52, 1995.
- Fuchs J, Cavdar S, Blumenstock G, et al.: POST-TEXT III and IV Hepatoblastoma: Extended Hepatic Resection Avoids Liver Transplantation in Selected Cases. Ann Surg 266 (2): 318-323, 2017.
- Hemming AW, Reed AI, Fujita S, et al.: Role for extending hepatic resection using an aggressive approach to liver surgery. J Am Coll Surg 206 (5): 870-5; discussion 875-8, 2008.
- Fonseca A, Gupta A, Shaikh F, et al.: Extreme hepatic resections for the treatment of advanced hepatoblastoma: Are planned close margins an acceptable approach? Pediatr Blood Cancer 65 (2): , 2018.
- de Freitas Paganoti G, Tannuri ACA, Dantas Marques AC, et al.: Extensive Hepatectomy as an Alternative to Liver Transplant in Advanced Hepatoblastoma: A New Protocol Used in a Pediatric Liver Transplantation Center. Transplant Proc 51 (5): 1605-1610, 2019.
- Katzenstein HM, Malogolowkin MH, Krailo MD, et al.: Doxorubicin in combination with cisplatin, 5-flourouracil, and vincristine is feasible and effective in unresectable hepatoblastoma: A Children's Oncology Group study. Cancer 128 (5): 1057-1065, 2022.
- Wang S, Yang C, Zhang J, et al.: First experience of high-intensity focused ultrasound combined with transcatheter arterial embolization as local control for hepatoblastoma. Hepatology 59 (1): 170-7, 2014.
- Meyers RL, Katzenstein HM, Krailo M, et al.: Surgical resection of pulmonary metastatic lesions in children with hepatoblastoma. J Pediatr Surg 42 (12): 2050-6, 2007.
- Karski EE, Dvorak CC, Leung W, et al.: Treatment of hepatoblastoma with high-dose chemotherapy and stem cell rescue: the pediatric blood and marrow transplant consortium experience and review of the literature. J Pediatr Hematol Oncol 36 (5): 362-8, 2014.
- Partrick DA, Bensard DD, Teitelbaum DH, et al.: Successful thoracoscopic lung biopsy in children utilizing preoperative CT-guided localization. J Pediatr Surg 37 (7): 970-3; discussion 970-3, 2002.
- Katzenstein HM, Rigsby C, Shaw PH, et al.: Novel therapeutic approaches in the treatment of children with hepatoblastoma. J Pediatr Hematol Oncol 24 (9): 751-5, 2002.
- Palmer RD, Williams DM: Dramatic response of multiply relapsed hepatoblastoma to irinotecan (CPT-11). Med Pediatr Oncol 41 (1): 78-80, 2003.
- Qayed M, Powell C, Morgan ER, et al.: Irinotecan as maintenance therapy in high-risk hepatoblastoma. Pediatr Blood Cancer 54 (5): 761-3, 2010.
- Zsíros J, Brugières L, Brock P, et al.: Efficacy of irinotecan single drug treatment in children with refractory or recurrent hepatoblastoma--a phase II trial of the childhood liver tumour strategy group (SIOPEL). Eur J Cancer 48 (18): 3456-64, 2012.
- Sue K, Ikeda K, Nakagawara A, et al.: Intrahepatic arterial injections of cisplatin-phosphatidylcholine-Lipiodol suspension in two unresectable hepatoblastoma cases. Med Pediatr Oncol 17 (6): 496-500, 1989.
- Habrand JL, Nehme D, Kalifa C, et al.: Is there a place for radiation therapy in the management of hepatoblastomas and hepatocellular carcinomas in children? Int J Radiat Oncol Biol Phys 23 (3): 525-31, 1992.
- Semeraro M, Branchereau S, Maibach R, et al.: Relapses in hepatoblastoma patients: clinical characteristics and outcome--experience of the International Childhood Liver Tumour Strategy Group (SIOPEL). Eur J Cancer 49 (4): 915-22, 2013.
- Shi Y, Geller JI, Ma IT, et al.: Relapsed hepatoblastoma confined to the lung is effectively treated with pulmonary metastasectomy. J Pediatr Surg 51 (4): 525-9, 2016.
- Matsunaga T, Sasaki F, Ohira M, et al.: Analysis of treatment outcome for children with recurrent or metastatic hepatoblastoma. Pediatr Surg Int 19 (3): 142-6, 2003.
- Malogolowkin MH, Katzenstein HM, Krailo M, et al.: Redefining the role of doxorubicin for the treatment of children with hepatoblastoma. J Clin Oncol 26 (14): 2379-83, 2008.
- Trobaugh-Lotrario AD, Meyers RL, Feusner JH: Outcomes of Patients With Relapsed Hepatoblastoma Enrolled on Children's Oncology Group (COG) Phase I and II Studies. J Pediatr Hematol Oncol 38 (3): 187-90, 2016.
- Yevich S, Calandri M, Gravel G, et al.: Reiterative Radiofrequency Ablation in the Management of Pediatric Patients with Hepatoblastoma Metastases to the Lung, Liver, or Bone. Cardiovasc Intervent Radiol 42 (1): 41-47, 2019.
- Liu B, Zhou L, Huang G, et al.: First Experience of Ultrasound-guided Percutaneous Ablation for Recurrent Hepatoblastoma after Liver Resection in Children. Sci Rep 5: 16805, 2015.
Carcinoma hepatocelular
Incidencia
La incidencia anual de carcinoma hepatocelular en los Estados Unidos es 0,4 por millón de niños de 0 a 14 años y de 1,4 por millón de adolescentes de 15 a 19 años. Aunque la incidencia de carcinoma hepatocelular en adultos en los Estados Unidos ha aumentado de manera constante desde la década de 1970, posiblemente debido al aumento de la frecuencia de infección crónica por el virus de la hepatitis C, la incidencia en los niños no ha aumentado. En varios países asiáticos, la incidencia de carcinoma hepatocelular en la niñez es 10 veces más alta que en América del Norte. La incidencia alta esta relacionada con la incidencia de hepatitis B adquirida en el periodo perinatal, que es prevenible en la mayoría de los casos mediante la vacunación y administración de inmunoglobulina antihepatitis B al recién nacido.
El carcinoma hepatocelular fibrolamelar, un subtipo de carcinoma hepatocelular que no está relacionado con cirrosis, infección por el virus de la hepatitis B (VHB) o de la hepatitis C (VHC), se presenta por lo general en adolescentes y adultos jóvenes, pero también se ha notificado en lactantes.
Factores de riesgo
Las afecciones que se relacionan con el carcinoma hepatocelular se describen en el Cuadro 7.
| Trastornos relacionados | Hallazgos clínicos |
|---|---|
| Síndrome de Alagille | Frente prominente y ancha, ojos hundidos y mentón pequeño saliente. La anomalía de las vías biliares produce cicatrización intrahepática. Para obtener más información, consultar la sección Síndrome de Alagille. |
| Enfermedades por almacenamiento de glucógeno I–IV | Los síntomas varían según el trastorno. |
| Hepatitis B y C | Para obtener más información, consultar la sección Infección por virus de la hepatitis B y de la hepatitis C. |
| Colestasis intrahepática familiar progresiva | Los síntomas de ictericia, prurito, y retraso del desarrollo comienzan en la lactancia y progresan hasta hipertensión portal e insuficiencia hepática. |
| Tirosinemia | Los primeros meses de vida: retraso del desarrollo, vómitos e ictericia. |
Síndrome de Alagille
El síndrome de Alagille es un síndrome genético autosómico dominante que suele obedecer a una mutación o deleción en el gen JAG1. Afecta las vías biliares hepáticas, el corazón y los vasos sanguíneos del encéfalo y el riñón. Los pacientes presentan una facies característica.
Infección por virus de la hepatitis B y de la hepatitis C
En los niños, el carcinoma hepatocelular se relaciona con la infección perinatal por el virus de la hepatitis B (VHB). En los adultos, se relaciona con infecciones crónicas por el VHB y el virus de la hepatitis C (VHC). La amplia diseminación de la vacunación contra la hepatitis B ha disminuido la incidencia del carcinoma hepatocelular en Asia. En comparación con los adultos, el período de incubación desde la infección por el virus de la hepatitis hasta la génesis del carcinoma hepatocelular es extremadamente corto en un subgrupo pequeño de niños con virus adquirido en el periodo perinatal. Las mutaciones en el gen MET podrían ser un mecanismo que acorte el período de incubación.
La infección por el virus de la hepatitis C se relaciona con cirrosis y carcinoma hepatocelular que tarda décadas en formarse y que, por lo general, no se observa en niños. A diferencia de los adultos, la cirrosis en los niños es un factor mucho menos frecuente en la presentación del carcinoma hepatocelular y solo se encuentra en un 20 % a un 35 % de los niños con esta enfermedad.
Lesión hepática no causada por virus
Los tipos específicos de lesiones hepáticas no causadas por virus y la cirrosis que se relacionan con el carcinoma hepatocelular en los niños son las siguientes:
- Tirosinemia. Los pacientes con tirosinemia se someten en forma regular a exámenes de detección de carcinoma hepatocelular, aunque se traten con nitisinona. La nitisinona puede prevenir la cirrosis y disminuir la incidencia del carcinoma hepatocelular, en especial cuando se administra durante la lactancia, después de los exámenes de detección neonatales para diagnosticar la tirosinemia. Hasta 2014, solo una minoría de los programas estatales de detección habían adoptado un examen de detección para recién nacidos nuevo, más predictivo y muy recomendado, que es mucho más eficaz en las primeras 24 a 48 horas de vida del bebé.
En un estudio realizado en Irán, 36 niños se sometieron a trasplante de hígado por tirosinemia. De estos niños, 22 tenían nódulos hepáticos mayores de 10 cm, y en 20 niños, los nódulos eran cirróticos. La mediana de edad en el momento del trasplante fue de 3,9 años. Se encontró carcinoma hepatocelular en el hígado resecado de 5 de los 19 niños mayores de 2 años y en ninguno de los niños menores de 2 años.
- Colestasis intrahepática familiar progresiva. El carcinoma hepatocelular también se presenta en niños muy pequeños con mutaciones en el gen ABCB11 (codificador de la proteína de la bomba exportadora de sales biliares), lo que causa colestasis intrahepática familiar progresiva.
Características genómicas del carcinoma hepatocelular
Características moleculares del carcinoma hepatocelular
Los hallazgos genómicos relacionados con el carcinoma hepatocelular son los siguientes:
- En un caso de carcinoma hepatocelular pediátrico analizado mediante secuenciación de exoma completo, se observó una tasa de mutación más alta (53 variantes) y la coexistencia de mutaciones en CTNNB1 y NFE2L2.
- En un estudio se investigaron tumores de carcinoma hepatocelular no fibrolamelar pediátrico (N = 15) mediante varios instrumentos analíticos. Se encontró que estos tumores a menudo portan alteraciones en un subgrupo de genes que por lo general están mutados en el carcinoma hepatocelular de adultos, como CTNNB1 y TERT, pero los mecanismos moleculares que dan origen a estas mutaciones son diferentes. La mutación en TP53 fue infrecuente en esta cohorte de carcinoma hepatocelular pediátrico. El carcinoma hepatocelular pediátrico que surge en un paciente con una enfermedad metabólica subyacente exhibe menos mutaciones y su perfil molecular es diferente. En este grupo de pacientes no se encontraron las mutaciones oncoiniciadoras características.
- El carcinoma hepatocelular fibrolamelar es un subtipo raro de carcinoma hepatocelular que se observa en niños más grandes y en adultos jóvenes. Se caracteriza por una deleción de casi 400 kB en el cromosoma 19 que deriva en la elaboración de un RNA híbrido que codifica una proteína que contiene el dominio aminoterminal de DNAJB1, un homólogo de la carabina molecular DNAJ, fundida en marco con PRKACA, el dominio catalítico de la proteína cinasa A.
- Un subtipo raro de cáncer de hígado infantil de mayor malignidad (neoplasia hepatocelular sin otra indicación, que también se llama tumor de células de transición de hígado) se presenta en niños mayores. Tiene características clínicas e histopatológicas de hepatoblastoma y de carcinoma hepatocelular.
Se observaron mutaciones en TERT en 2 de los 4 casos analizados de tumor de células de transición de hígado. Las mutaciones en TERT también se observaron con frecuencia en adultos con carcinoma hepatocelular.
Hasta la fecha, no se utilizan estas mutaciones genéticas en la selección de fármacos para ensayos clínicos de investigación.
Diagnóstico
Para obtener más información, consultar la sección Diagnóstico en la sección Hepatoblastoma.
Pronóstico y factores pronósticos
Pronóstico
La tasa de supervivencia general (SG) a 5 años es del 42 % en los niños y adolescentes con carcinoma hepatocelular. La supervivencia a 5 años de los pacientes con carcinoma hepatocelular depende del estadio de la enfermedad. En un estudio intergrupal de quimioterapia realizado en la década de 1990, 7 de 8 pacientes en estadio I sobrevivieron y menos del 10 % de los pacientes en estadio III y IV sobrevivieron. En un análisis de los datos del Surveillance, Epidemiology, and End Results (SEER) Program, se observó una tasa de SG a 5 años del 24 %, una tasa de SG a 10 años del 23 % y una tasa de SG a 20 años del 8 % en pacientes de 19 años o menos; lo que indica una mejora de los desenlaces con el tratamiento más reciente. En un análisis multivariante de datos del SEER, la resección quirúrgica, el tumor localizado y una etnia diferente a la hispana se relacionaron con una mejora del desenlace. Los pacientes que se sometieron a una resección quirúrgica completa tuvieron una tasa de SG del 60 %, en comparación con una tasa de SG del 0 % en los pacientes que se sometieron a una resección incompleta.[Nivel de evidencia C1]
Se encontró que las tasas de SG a 5 años según el grupo PRE-Treatment EXTent of disease (PRETEXT) en los pacientes con carcinoma hepatocelular en el ensayo SIOPEL-1 fueron las siguientes:
- 44 % en pacientes con tumores del grupo PRETEXT I.
- 44 % en pacientes con tumores del grupo PRETEXT II.
- 22 % en pacientes con tumores del grupo PRETEXT III.
- 8 % en pacientes con tumores del grupo PRETEXT IV.
Para obtener más información sobre la agrupación de PRETEXT, consultar la sección Definiciones de los grupos PRETEXT y POSTTEXT.
Pronóstico del carcinoma hepatocelular según el estadio quirúrgico de Evans Para obtener más información, consultar la sección Estadificación quirúrgica de Evans para el cáncer de hígado infantil (histórica).
- Estadio I.
Los niños con carcinoma hepatocelular en estadio I tienen un buen desenlace.
- Estadio II.
El estadio II se ve con muy poca frecuencia como para predecir el desenlace.
- Estadios III y IV
Los estadios III y IV suelen ser mortales.
Factores pronósticos
Los factores que afectan el pronóstico son los siguientes:
- Factores relacionados con el tratamiento: la cura del carcinoma hepatocelular exige una resección del tumor macroscópico. Sin embargo, el carcinoma hepatocelular con frecuencia invade de forma extensa o es multicéntrico, y menos del 30 % de los tumores son resecables. El trasplante ortotópico de hígado ha sido exitoso en niños seleccionados con carcinoma hepatocelular.
- Grupo PRETEXT: el grupo PRETEXT (resecabilidad) también es un factor pronóstico. Para obtener más información, consultar la sección Estratificación tumoral por imágenes y estadificación quirúrgica de Evans para el cáncer de hígado infantil.
- Subtipo histológico del tumor: para obtener más información, consultar la sección Características histológicas.
Características histológicas
Las células del carcinoma hepatocelular tienen apariencia epitelial. El carcinoma hepatocelular por lo general surge en el lóbulo derecho del hígado.
Carcinoma fibrolamelar
Se ha descrito una variante histológica distintiva del carcinoma hepatocelular, denominada carcinoma fibrolamelar, en el hígado de niños mayores, adultos jóvenes y, en raras ocasiones, lactantes. Este tipo histológico se caracteriza por un transcrito de fusión creado por la deleción de una sección de 400 kb del cromosoma 19, que se encontró en 15 de 15 tumores que se analizaron.
A diferencia del carcinoma hepatocelular no fibrolamelar en adultos, la incidencia del carcinoma hepatocelular fibrolamelar en niños mayores y adultos no está aumentando de forma clara con el tiempo. El carcinoma fibrolamelar no se relaciona con cirrosis y se pensaba que tenía un mejor pronóstico. En estudios más antiguos, es posible que los mejores desenlaces en pacientes de carcinoma fibrolamelar se relacionaran con una proporción más alta de tumores menos invasores y más resecables en ausencia de cirrosis. Sin embargo, los desenlaces de los pacientes con carcinoma fibrolamelar en estudios prospectivos recientes, cuando se comparan estadio con estadio y grupo PRETEXT con grupo PRETEXT, son iguales a los desenlaces de los pacientes con carcinomas hepatocelulares convencionales.; [Nivel de evidencia C1]
Neoplasia hepatocelular sin otra indicación
La neoplasia hepatocelular sin otra indicación (SAI) también se conoce como tumor de células de transición de hígado. Este tumor, con características de hepatoblastoma y carcinoma hepatocelular es una neoplasia rara que se presenta en niños mayores y adolescentes Las células tiene una posición intermedia entre los hepatoblastos y las células tumorales más maduras parecidas a hepatocitos. Las células tumorales a veces varían en diferentes regiones del tumor entre un hepatoblastoma clásico y un carcinoma hepatocelular obvio. En la clasificación internacional de consenso, estos tumores se llaman neoplasia hepatocelular, SAI. Por lo general, los tumores son unifocales y a veces exhiben necrosis central en el momento de la presentación inicial. No se ha estudiado rigurosamente la respuesta a la quimioterapia, pero se cree que es similar a la respuesta del carcinoma hepatocelular.
childhood hepatocellular carcinomaTratamiento del carcinoma hepatocelular
Las opciones de tratamiento del carcinoma hepatocelular recién diagnosticado dependen de los siguientes aspectos:
- La resecabilidad del cáncer en el momento del diagnóstico.
- La respuesta del cáncer a la quimioterapia.
- La presencia de metástasis del cáncer.
- La relación del cáncer con el virus de la hepatitis B.
Opciones de tratamiento del carcinoma hepatocelular resecable en el momento del diagnóstico
Las opciones de tratamiento del carcinoma hepatocelular resecable en el momento del diagnóstico son las siguientes:
- Resección quirúrgica completa del tumor primario seguida de quimioterapia.
- Quimioterapia seguida de resección quirúrgica completa del tumor primario.
- Resección quirúrgica completa sin quimioterapia.
La quimioterapia y la resección quirúrgica son los pilares del tratamiento del carcinoma hepatocelular resecable.
Evidencia (resección quirúrgica completa seguida de quimioterapia):
- Entre 8 pacientes con carcinoma hepatocelular en estadio I que recibieron quimioterapia adyuvante a base de cisplatino, 7 sobrevivieron sin enfermedad.
- En una encuesta sobre tumores de hígado infantil tratados antes del uso sistemático de quimioterapia, solo sobrevivieron 12 de 33 pacientes con carcinoma hepatocelular sometidos a escisión completa del tumor. Esto indica que el tratamiento con quimioterapia adyuvante puede beneficiar a los niños con carcinoma hepatocelular completamente resecado.
- En un análisis de los datos de SEER para niños y adolescentes menores de 20 años diagnosticados entre 1976 y 2009, los pacientes que se sometieron a una resección completa tuvieron una tasa de SG a 5 años del 60 %, y los pacientes que no se sometieron a una resección completa tuvieron una tasa de SG a 5 años del 0 %.[Nivel de evidencia C1]
Es posible administrar cisplatino y doxorrubicina como terapia adyuvante porque estos fármacos pueden tener actividad para el tratamiento del carcinoma hepatocelular.
Evidencia (resección quirúrgica completa sin quimioterapia):
- En un informe retrospectivo de una sola institución, 12 pacientes con carcinoma hepatocelular en estadio I se trataron con cirugía. De estos pacientes, 10 no recibieron quimioterapia y 2 recibieron un ciclo corto de quimioterapia según la preferencia del oncólogo.[Nivel de evidencia C1]
- Los 12 pacientes permanecieron vivos sin indicios de enfermedad al cabo de una mediana de seguimiento de 54 meses.
A pesar de las mejoras de los últimos 20 años en las técnicas quirúrgicas, la administración de quimioterapia y los cuidados de apoyo para los pacientes, en los ensayos clínicos de quimioterapia contra el cáncer no se han observado mejoras en las tasas de supervivencia de pacientes pediátricos con carcinoma hepatocelular. En los estudios del International Childhood Liver Tumors Strategy Group (SIOPEL) en Europa, no se han observado mejoras en la SG a 5 años desde 1990. Los únicos sobrevivientes a largo plazo fueron pacientes con tumores resecables en el momento del diagnóstico, que fueron menos del 30 % de los niños inscritos en el estudio. Sin embargo, en algunos estudios de trasplante de hígado (resección completa con trasplante y quimioterapia neoadyuvante o sin esta) se observaron tasas de SG superiores a las de los estudios SIOPEL.
Opciones de tratamiento del carcinoma hepatocelular no metastásico que no es resecable en el momento del diagnóstico
Las opciones de tratamiento del carcinoma hepatocelular no metastásico que no es resecable en el momento del diagnóstico son las siguientes:
- Quimioterapia seguida de una nueva evaluación de la resecabilidad quirúrgica. Si el tumor primario es resecable, resección quirúrgica completa.
- Quimioterapia con radioembolización transarterial (RETA) o sin esta, seguida de una nueva evaluación de la resecabilidad quirúrgica. Si el tumor primario permanece irresecable:
- Trasplante ortotópico de hígado.
- Quimioembolización transarterial (QETA) o RETA seguida de resección completa o trasplante de hígado.
- QETA o RETA solas.
Para curar este cáncer, es necesaria la administración de quimioterapia o QETA neoadyuvantes a fin de mejorar la resecabilidad o el trasplante de hígado, lo que podría permitir la resección completa del tumor.
Evidencia (quimioterapia seguida de cirugía):
- En un estudio prospectivo de 41 pacientes que recibieron quimioterapia preoperatoria con cisplatino y doxorrubicina, se observó lo siguiente:
- El tratamiento produjo una disminución del tumor, con reducción de las concentraciones de alfafetoproteína (AFP) en cerca del 50 % de los pacientes.
- Quienes respondieron al tratamiento tuvieron resecabilidad del tumor y supervivencia superiores, aunque la tasa de SG fue del 28 % y solo sobrevivieron aquellos que se sometieron a resección completa.
Evidencia (quimioterapia, RETA o QETA seguidas de una nueva evaluación de la resecabilidad quirúrgica; opciones de tratamiento, incluso trasplante de hígado, para el tumor primario irresecable luego de quimioterapia, RETA o QETA):
- El trasplante de hígado ha sido un tratamiento exitoso para los niños con carcinoma hepatocelular irresecable. La tasa de supervivencia es de alrededor del 60 %, y la mayoría de las muertes se deben a recidiva tumoral.
- En una revisión de los datos de SEER para el tratamiento del carcinoma hepatocelular en pacientes menores de 20 años, se reveló que el 75 % de los pacientes se sometieron a resección y el 25 % a trasplante de hígado.
- La tasa de SG a 5 años fue del 53,4 % con la resección y del 85,3 % con el trasplante, lo que indica que los criterios de trasplante para el carcinoma hepatocelular se podrían flexibilizar para el beneficio general de los pacientes. Estos datos no se han verificado en un ensayo clínico prospectivo.
- La QETA seguida de resección quirúrgica completa del tumor primario quizás sea una alternativa al uso de quimioterapia seguida de resección quirúrgica.
- En estudios realizados en adultos en China, se indica que la repetición de la QETA hepática antes de la cirugía en ocasiones mejora el desenlace de la hepatectomía posterior.
- En un metanálisis se encontraron 7 ensayos aleatorizados en los que se comparó la resección sola con la QETA seguida de resección. No hubo diferencia en la supervivencia sin complicaciones (SSC) a 3 años y la SG entre los dos grupos, pero la SSC a 5 años y la SG favorecieron el uso de QETA seguida de resección.
- La RETA se ha utilizado para el tratamiento de pacientes adultos con carcinoma hepatocelular durante un tiempo. En un número pequeño de pacientes, la RETA proporcionó tanto un beneficio paliativo como un posible puente para el trasplante de hígado.
Si el tumor primario no es resecable luego de la quimioterapia y el paciente no es apto para un trasplante, los otros abordajes de tratamiento que se usan en adultos son los siguientes:
- Sorafenib.
- QETA o RETA.
- Criocirugía.
- Inyección intratumoral de alcohol.
- Radioterapia.
Hay pocos datos sobre el uso de estos abordajes de tratamiento en niños.
En los pocos datos provenientes de un estudio piloto realizado en Europa se indica que 12 niños y adolescentes con diagnóstico reciente de carcinoma hepatocelular en estadio avanzado toleraron bien el sorafenib cuando se administró en combinación con quimioterapia estándar de cisplatino y doxorrubicina. Se necesita más estudios para definir su función en el tratamiento de los niños con carcinoma hepatocelular.
La criocirugía, la inyección intratumoral de alcohol y la ablación por radiofrecuencia pueden tratar con éxito tumores pequeños (<5 cm) en adultos con hígado cirrótico. Algunos abordajes locales como la criocirugía, la ablación por radiofrecuencia y la QETA, que suprimen la progresión tumoral del carcinoma hepatocelular, se usan como tratamiento puente en los adultos con el fin de retrasar el crecimiento del tumor mientras están en lista de espera de un trasplante de hígado cadavérico. En un estudio pediátrico de 8 pacientes con carcinoma hepatocelular, 2 pacientes murieron por enfermedad progresiva sin trasplante. El tratamiento con TACE estabilizó la enfermedad en 6 pacientes durante una media de 141 días hasta llegar al trasplante.[Nivel de evidencia C1] De ellos, 5 pacientes estaban vivos al final del período de observación y un paciente murió por enfermedad. Para obtener más información, consultar Tratamiento del cáncer primario de hígado.
Opciones de tratamiento del carcinoma hepatocelular con metástasis en el momento del diagnóstico
Ningún tratamiento específico ha sido eficaz para tratar el carcinoma hepatocelular metastásico en el grupo de edad pediátrica.
En dos ensayos prospectivos, el cisplatino con vincristina o fluorouracilo o la infusión continua de doxorrubicina no fueron eficaces para tratar de manera adecuada a 25 pacientes con carcinoma hepatocelular metastásico. Algunos pacientes quizás se beneficien de manera transitoria del tratamiento con cisplatino o doxorrubicina, en particular, si el tumor hepático localizado se reduce de manera adecuada para permitir la resección de la enfermedad, y la enfermedad metastásica desaparece o se torna resecable.
Opciones de tratamiento del carcinoma hepatocelular relacionado con el virus de la hepatitis B
Aunque el carcinoma hepatocelular relacionado con el virus de la hepatitis B (VHB) no es común en los niños en los Estados Unidos, se sabe que el tratamiento con nucleótido o análogo nucleósido del inhibidor del VHB mejora el pronóstico posoperatorio en niños y adultos tratados en China.
La opción de tratamiento del carcinoma hepatocelular relacionado con el virus de la hepatitis B es la siguiente:
- Terapia antiviral.
Evidencia (terapia antiviral):
- En un ensayo controlado aleatorizado, se evaluó la respuesta de 163 pacientes después de una hepatectomía radical a uno de 3 tratamientos antivirales.
- En un análisis multivariante de Cox, se observó que la terapia antiviral disminuyó de manera significativa la recidiva del carcinoma hepatocelular, con un cociente de riesgos instantáneos (CRI) de 0,48 (intervalo de confianza [IC] 95 %, 0,32–0,70) así como las muertes relacionadas con el carcinoma hepatocelular, con un CRI de 0,26 (IC 95 %, 0,14–0,50).
- Los pacientes que recibieron terapia antiviral exhibieron una reducción significativa de la recidiva temprana (CRI 0,41; IC 95 % 0,27–0,62) y una mejoría del funcionamiento hepático 6 meses después de la cirugía en comparación con los controles (P< 0,001).
Tratamiento del carcinoma hepatocelular progresivo o recidivante
El pronóstico de un paciente con carcinoma hepatocelular recidivante o progresivo es muy precario.
La opción de tratamiento del carcinoma hepatocelular progresivo o recidivante es la siguiente:
- Temporización de la quimioembolización antes del trasplante o inmediatamente después del trasplante de hígado, para aquellos con recidiva aislada en el hígado.
- Es posible que los ensayos clínicos de fase l y fase II sean apropiados y se deben considerar.
El tratamiento con sorafenib produjo una mejora de la supervivencia sin progresión en adultos con carcinoma hepatocelular en estadio avanzado. En los pacientes adultos que recibieron sorafenib, la mediana de supervivencia y el tiempo hasta la progresión radiológica fueron alrededor de 3 meses más largos que en los pacientes que recibieron un placebo.
childhood hepatocellular carcinomaOpciones de tratamiento en evaluación clínica para el carcinoma hepatocelular
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de ensayo clínico nacional o institucional en curso:
- AHEP1531 (NCT03533582) (Cisplatin and Combination Chemotherapy in Treating Children and Young Adults with Hepatoblastoma or Liver Cancer After Surgery):
este ensayo representa la participación del COG en un gran ensayo internacional (Pediatric Hepatic Malignancy International Therapeutic Trial [PHITT]) de tratamiento para todos los estadios de hepatoblastoma y carcinoma hepatocelular en niños.
- El carcinoma hepatocelular potencialmente resecable se trata con resección completa sin quimioterapia si la masa parece obedecer a una enfermedad hepática subyacente. Si es un carcinoma hepatocelular nuevo sin enfermedad subyacente, se trata con resección seguida de 4 ciclos de cisplatino y doxorrubicina.
- Los pacientes con carcinoma hepatocelular metastásico o irresecable en el momento del diagnóstico se derivan a consulta con los servicios de radiología intervencionista y trasplante de hígado. Luego los pacientes se asignan al azar a recibir cisplatino, doxorrubicina y sorafenib durante 3 ciclos de 21 días, o cisplatino, doxorrubicina y sorafenib alternados con gemcitabina, oxaliplatino y sorafenib durante 4 ciclos de 14 días. Los pacientes que respondieron continúan con la quimioterapia durante el mismo número de ciclos (3 o 4). Reciben el mismo régimen de quimioterapia asignado originalmente.
References
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 15, 2023.
- El-Serag HB, Davila JA, Petersen NJ, et al.: The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med 139 (10): 817-23, 2003.
- Chang MH, Chen TH, Hsu HM, et al.: Prevention of hepatocellular carcinoma by universal vaccination against hepatitis B virus: the effect and problems. Clin Cancer Res 11 (21): 7953-7, 2005.
- Cruz O, Laguna A, Vancells M, et al.: Fibrolamellar hepatocellular carcinoma in an infant and literature review. J Pediatr Hematol Oncol 30 (12): 968-71, 2008.
- Keeffe EB, Pinson CW, Ragsdale J, et al.: Hepatocellular carcinoma in arteriohepatic dysplasia. Am J Gastroenterol 88 (9): 1446-9, 1993.
- Siciliano M, De Candia E, Ballarin S, et al.: Hepatocellular carcinoma complicating liver cirrhosis in type IIIa glycogen storage disease. J Clin Gastroenterol 31 (1): 80-2, 2000.
- Ni YH, Chang MH, Hsu HY, et al.: Hepatocellular carcinoma in childhood. Clinical manifestations and prognosis. Cancer 68 (8): 1737-41, 1991.
- Tsukuma H, Hiyama T, Tanaka S, et al.: Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med 328 (25): 1797-801, 1993.
- González-Peralta RP, Langham MR, Andres JM, et al.: Hepatocellular carcinoma in 2 young adolescents with chronic hepatitis C. J Pediatr Gastroenterol Nutr 48 (5): 630-5, 2009.
- Knisely AS, Strautnieks SS, Meier Y, et al.: Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 44 (2): 478-86, 2006.
- Alonso EM, Snover DC, Montag A, et al.: Histologic pathology of the liver in progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr 18 (2): 128-33, 1994.
- van Spronsen FJ, Bijleveld CM, van Maldegem BT, et al.: Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifluoro- methylbenzoyl)-1, 3-cyclohexanedione treatment. J Pediatr Gastroenterol Nutr 40 (1): 90-3, 2005.
- Park WS, Dong SM, Kim SY, et al.: Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer Res 59 (2): 307-10, 1999.
- de Laet C, Dionisi-Vici C, Leonard JV, et al.: Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis 8: 8, 2013.
- De Jesús VR, Adam BW, Mandel D, et al.: Succinylacetone as primary marker to detect tyrosinemia type I in newborns and its measurement by newborn screening programs. Mol Genet Metab 113 (1-2): 67-75, 2014 Sep-Oct.
- Bahador A, Dehghani SM, Geramizadeh B, et al.: Liver Transplant for Children With Hepatocellular Carcinoma and Hereditary Tyrosinemia Type 1. Exp Clin Transplant 13 (4): 329-32, 2015.
- Vilarinho S, Erson-Omay EZ, Harmanci AS, et al.: Paediatric hepatocellular carcinoma due to somatic CTNNB1 and NFE2L2 mutations in the setting of inherited bi-allelic ABCB11 mutations. J Hepatol 61 (5): 1178-83, 2014.
- Haines K, Sarabia SF, Alvarez KR, et al.: Characterization of pediatric hepatocellular carcinoma reveals genomic heterogeneity and diverse signaling pathway activation. Pediatr Blood Cancer 66 (7): e27745, 2019.
- Honeyman JN, Simon EP, Robine N, et al.: Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 343 (6174): 1010-4, 2014.
- Eichenmüller M, Trippel F, Kreuder M, et al.: The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J Hepatol 61 (6): 1312-20, 2014.
- Nault JC, Mallet M, Pilati C, et al.: High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun 4: 2218, 2013.
- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations). National Cancer Institute, 2012, Section 29. Also available online. Last accessed August 11, 2022.
- Katzenstein HM, Krailo MD, Malogolowkin MH, et al.: Hepatocellular carcinoma in children and adolescents: results from the Pediatric Oncology Group and the Children's Cancer Group intergroup study. J Clin Oncol 20 (12): 2789-97, 2002.
- Allan BJ, Wang B, Davis JS, et al.: A review of 218 pediatric cases of hepatocellular carcinoma. J Pediatr Surg 49 (1): 166-71; discussion 171, 2014.
- Czauderna P, Mackinlay G, Perilongo G, et al.: Hepatocellular carcinoma in children: results of the first prospective study of the International Society of Pediatric Oncology group. J Clin Oncol 20 (12): 2798-804, 2002.
- Douglass E, Ortega J, Feusner J, et al.: Hepatocellular carcinoma (HCA) in children and adolescents: results from the Pediatric Intergroup Hepatoma Study (CCG 8881/POG 8945). [Abstract] Proceedings of the American Society of Clinical Oncology 13: A-1439, 420, 1994.
- Austin MT, Leys CM, Feurer ID, et al.: Liver transplantation for childhood hepatic malignancy: a review of the United Network for Organ Sharing (UNOS) database. J Pediatr Surg 41 (1): 182-6, 2006.
- Vinayak R, Cruz RJ, Ranganathan S, et al.: Pediatric liver transplantation for hepatocellular cancer and rare liver malignancies: US multicenter and single-center experience (1981-2015). Liver Transpl 23 (12): 1577-1588, 2017.
- Eggert T, McGlynn KA, Duffy A, et al.: Fibrolamellar hepatocellular carcinoma in the USA, 2000-2010: A detailed report on frequency, treatment and outcome based on the Surveillance, Epidemiology, and End Results database. United European Gastroenterol J 1 (5): 351-7, 2013.
- Mayo SC, Mavros MN, Nathan H, et al.: Treatment and prognosis of patients with fibrolamellar hepatocellular carcinoma: a national perspective. J Am Coll Surg 218 (2): 196-205, 2014.
- Katzenstein HM, Krailo MD, Malogolowkin MH, et al.: Fibrolamellar hepatocellular carcinoma in children and adolescents. Cancer 97 (8): 2006-12, 2003.
- Weeda VB, Murawski M, McCabe AJ, et al.: Fibrolamellar variant of hepatocellular carcinoma does not have a better survival than conventional hepatocellular carcinoma--results and treatment recommendations from the Childhood Liver Tumour Strategy Group (SIOPEL) experience. Eur J Cancer 49 (12): 2698-704, 2013.
- López-Terrada D, Alaggio R, de Dávila MT, et al.: Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol 27 (3): 472-91, 2014.
- Prokurat A, Kluge P, Kościesza A, et al.: Transitional liver cell tumors (TLCT) in older children and adolescents: a novel group of aggressive hepatic tumors expressing beta-catenin. Med Pediatr Oncol 39 (5): 510-8, 2002.
- Exelby PR, Filler RM, Grosfeld JL: Liver tumors in children in the particular reference to hepatoblastoma and hepatocellular carcinoma: American Academy of Pediatrics Surgical Section Survey--1974. J Pediatr Surg 10 (3): 329-37, 1975.
- D'Souza AM, Shah R, Gupta A, et al.: Surgical management of children and adolescents with upfront completely resected hepatocellular carcinoma. Pediatr Blood Cancer 65 (11): e27293, 2018.
- Murawski M, Weeda VB, Maibach R, et al.: Hepatocellular Carcinoma in Children: Does Modified Platinum- and Doxorubicin-Based Chemotherapy Increase Tumor Resectability and Change Outcome? Lessons Learned From the SIOPEL 2 and 3 Studies. J Clin Oncol 34 (10): 1050-6, 2016.
- Kelly D, Sharif K, Brown RM, et al.: Hepatocellular carcinoma in children. Clin Liver Dis 19 (2): 433-47, 2015.
- Malek MM, Shah SR, Atri P, et al.: Review of outcomes of primary liver cancers in children: our institutional experience with resection and transplantation. Surgery 148 (4): 778-82; discussion 782-4, 2010.
- Ismail H, Broniszczak D, Kaliciński P, et al.: Liver transplantation in children with hepatocellular carcinoma. Do Milan criteria apply to pediatric patients? Pediatr Transplant 13 (6): 682-92, 2009.
- Pham TA, Gallo AM, Concepcion W, et al.: Effect of Liver Transplant on Long-term Disease-Free Survival in Children With Hepatoblastoma and Hepatocellular Cancer. JAMA Surg 150 (12): 1150-8, 2015.
- Reyes JD, Carr B, Dvorchik I, et al.: Liver transplantation and chemotherapy for hepatoblastoma and hepatocellular cancer in childhood and adolescence. J Pediatr 136 (6): 795-804, 2000.
- Bilik R, Superina R: Transplantation for unresectable liver tumors in children. Transplant Proc 29 (7): 2834-5, 1997.
- Romano F, Stroppa P, Bravi M, et al.: Favorable outcome of primary liver transplantation in children with cirrhosis and hepatocellular carcinoma. Pediatr Transplant 15 (6): 573-9, 2011.
- McAteer JP, Goldin AB, Healey PJ, et al.: Surgical treatment of primary liver tumors in children: outcomes analysis of resection and transplantation in the SEER database. Pediatr Transplant 17 (8): 744-50, 2013.
- Zhang Z, Liu Q, He J, et al.: The effect of preoperative transcatheter hepatic arterial chemoembolization on disease-free survival after hepatectomy for hepatocellular carcinoma. Cancer 89 (12): 2606-12, 2000.
- Yu T, Xu X, Chen B: TACE combined with liver resection versus liver resection alone in the treatment of resectable HCC: a meta-analysis. Chinese-German J Clin Oncol 12 (11): 532-6, 2013.
- Tohme S, Bou Samra P, Kaltenmeier C, et al.: Radioembolization for Hepatocellular Carcinoma: A Nationwide 10-Year Experience. J Vasc Interv Radiol 29 (7): 912-919.e2, 2018.
- Aguado A, Ristagno R, Towbin AJ, et al.: Transarterial radioembolization with yttrium-90 of unresectable primary hepatic malignancy in children. Pediatr Blood Cancer 66 (7): e27510, 2019.
- Schmid I, Häberle B, Albert MH, et al.: Sorafenib and cisplatin/doxorubicin (PLADO) in pediatric hepatocellular carcinoma. Pediatr Blood Cancer 58 (4): 539-44, 2012.
- Zhou XD, Tang ZY: Cryotherapy for primary liver cancer. Semin Surg Oncol 14 (2): 171-4, 1998.
- Lencioni RA, Allgaier HP, Cioni D, et al.: Small hepatocellular carcinoma in cirrhosis: randomized comparison of radio-frequency thermal ablation versus percutaneous ethanol injection. Radiology 228 (1): 235-40, 2003.
- Lubienski A: Hepatocellular carcinoma: interventional bridging to liver transplantation. Transplantation 80 (1 Suppl): S113-9, 2005.
- Weiss KE, Sze DY, Rangaswami AA, et al.: Transarterial chemoembolization in children to treat unresectable hepatocellular carcinoma. Pediatr Transplant 22 (4): e13187, 2018.
- Yin J, Li N, Han Y, et al.: Effect of antiviral treatment with nucleotide/nucleoside analogs on postoperative prognosis of hepatitis B virus-related hepatocellular carcinoma: a two-stage longitudinal clinical study. J Clin Oncol 31 (29): 3647-55, 2013.
- Malogolowkin MH, Stanley P, Steele DA, et al.: Feasibility and toxicity of chemoembolization for children with liver tumors. J Clin Oncol 18 (6): 1279-84, 2000.
- Otte JB, Pritchard J, Aronson DC, et al.: Liver transplantation for hepatoblastoma: results from the International Society of Pediatric Oncology (SIOP) study SIOPEL-1 and review of the world experience. Pediatr Blood Cancer 42 (1): 74-83, 2004.
- Llovet JM, Ricci S, Mazzaferro V, et al.: Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359 (4): 378-90, 2008.
Sarcoma embrionario indiferenciado de hígado
Incidencia
El sarcoma embrionario indiferenciado de hígado (SEIH) es una entidad clínica y patológica distinta y representa entre el 2 % y el 15 % de las neoplasias malignas hepáticas en pediatría.
Diagnóstico
El SEIH se presenta como una masa abdominal, a menudo con dolor o malestar, por lo general, entre los 5 y 10 años de edad. La infiltración generalizada del hígado y las metástasis pulmonares son frecuentes Suele observarse como una neoplasia sólida o quística en las imágenes y, con frecuencia, hay necrosis central. Los sarcomas indiferenciados, como los hepatoblastomas indiferenciados de células pequeñas, se deben examinar mediante pruebas inmunohistoquímicas para detectar la pérdida de la expresión de SMARCB1 y ayudar a descartar tumores rabdoides hepáticos.
Es importante establecer la diferencia diagnóstica entre el SEIH y el rabdomiosarcoma de vías biliares porque, si bien comparten ciertas características clínicopatológicas, el tratamiento difiere como se muestra en el Cuadro 8. Para obtener más información, consultar Tratamiento del rabdomiosarcoma infantil.
| Sarcoma embrionario indiferenciado de hígado | Rabdomiosarcoma de vías biliares | |
|---|---|---|
| aAdaptación de Nicol et al. | ||
| Edad en el momento del diagnóstico | Mediana de edad, 10,5 años | Mediana de edad, 3,4 años |
| Localización del tumor | Con frecuencia surge en el lóbulo derecho del hígado | Con frecuencia surge en el hilio del hígado |
| Obstrucción de las vías biliares | Infrecuente | Frecuente; la ictericia es un síntoma común en el cuadro clínico inicial |
| Tratamiento | Cirugía y quimioterapia | Cirugía (por lo general, biopsia sola), radioterapia y quimioterapia |
Características histológicas
Las características histológicas distintivas del SEIH incluyen glóbulos hialinos intracelulares y anaplasia marcada sobre un fondo mesenquimatoso. Muchos tumores SEIH contienen diversos elementos de maduración de células mesenquimatosas, como del músculo liso y la grasa.
Hay evidencia histológica y clínica bien fundamentada que indica que el SEIH puede surgir dentro de hamartomas mesenquimatosos preexistentes en el hígado; estos son masas multiquísticas grandes y benignas que se presentan en los primeros 2 años de vida. En un informe de 11 casos de SEIH, 5 se presentaron en relación con hamartomas mesenquimatosos hepáticos y se observaron zonas de transición entre los subtipos histológicos. Muchos hamartomas mesenquimatosos de hígado exhiben una translocación característica con un punto de ruptura en 19q13.4, y varios SEIH tienen la misma translocación. Algunos SEIH que surgen de hamartomas mesenquimatosos de hígado tienen cariotipos complejos que no comprometen 19q13.4.
Pronóstico y factores pronósticos
Las tasas de supervivencia general (SG) de los niños con SEIH son superiores al 50 % cuando se combinan diferentes informes, aunque todas las series son pequeñas y es posible que tengan sesgo de selección.; [Nivel de evidencia C1]
En la Childhood Cancer Database, que no proporciona una revisión central de las características patológicas o detalles confiables del tratamiento no quirúrgico, se notificaron 103 niños con SEIH diagnosticado entre 1998 y 2012. La tasa de SG a 5 años de todos los pacientes fue del 86 % y en aquellos tratados con la combinación de cirugía y quimioterapia fue del 92 %. En un análisis multivariante de los datos no quirúrgicos, se observaron desenlaces más precarios estadísticamente significativos en los pacientes con tumores de más de 15 cm. De 10 niños, 7 presentaron metástasis y los 10 niños que se sometieron a trasplante ortotópico de hígado sobrevivieron por lo menos 5 años, pero no se describió el tratamiento en detalle.
Opciones de tratamiento del sarcoma embrionario indiferenciado de hígado
El sarcoma embrionario indiferenciado de hígado (SEIH) es raro. Solo se han publicado series pequeñas con respecto al tratamiento.
Las opciones de tratamiento del SEIH son las siguientes:
- Resección quirúrgica y quimioterapia.
- Trasplante de hígado para los tumores irresecables.
El abordaje aceptado en forma general, cuando es factible, es la resección de la masa tumoral primaria en el hígado. El uso de regímenes quimioterapéuticos intensivos ha mejorado la SG de los pacientes con SEIH. La quimioterapia neoadyuvante puede ser eficaz para disminuir el tamaño de una masa tumoral primaria irresecable, lo que resulta en resecabilidad. La mayoría de los pacientes se tratan con los regímenes quimioterapéuticos que se usan para el rabdomiosarcoma infantil o el sarcoma de Ewing, pero sin usar cisplatino.; [Nivel de evidencia C1]
Evidencia (resección quirúrgica y quimioterapia):
- Las únicas series prospectivas de tratamiento de pacientes con SEIH, estuvieron a cargo de los grupos cooperativos de sarcoma de tejido blando en Italia y Alemania. Los pacientes se trataron con cirugía conservadora o biopsia seguida de quimioterapia neoadyuvante compuesta de distintas combinaciones de vincristina, ciclofosfamida, dactinomicina, doxorrubicina e ifosfamida. La evaluación de la enfermedad, por lo general después de 4 ciclos de quimioterapia, siguió por una cirugía de segunda revisión cuando fue apropiado para tratar de extirpar el tumor primario residual, seguida de quimioterapia adicional o adyuvante.
- De 17 pacientes, 10 sobrevivieron en su primera remisión completa, y 1 paciente sobrevivió en su tercera remisión completa.
- En un informe retrospectivo de un solo centro, se informó que 5 pacientes con SEIH se trataron con cirugía y quimioterapia adyuvante que contenía vincristina, doxorrubicina, ciclofosfamida, ifosfamida y etopósido. De los pacientes, 4 tenían enfermedad en estadio I y 1 paciente tenía enfermedad en estadio II. Un paciente recibió radiación abdominal por ruptura tumoral.[Nivel de evidencia C1]
- Todos los pacientes están vivos (intervalo, 5–19 años), con tasas de supervivencia sin complicaciones y SG del 100 %.
En ocasiones, el trasplante de hígado se ha utilizado para tratar con éxito un tumor primario irresecable.
Opciones de tratamiento en evaluación clínica para el sarcoma embrionario indiferenciado de hígado
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
References
- Nicol K, Savell V, Moore J, et al.: Distinguishing undifferentiated embryonal sarcoma of the liver from biliary tract rhabdomyosarcoma: a Children's Oncology Group study. Pediatr Dev Pathol 10 (2): 89-97, 2007 Mar-Apr.
- Stocker JT: Hepatic tumors in children. Clin Liver Dis 5 (1): 259-81, viii-ix, 2001.
- Shehata BM, Gupta NA, Katzenstein HM, et al.: Undifferentiated embryonal sarcoma of the liver is associated with mesenchymal hamartoma and multiple chromosomal abnormalities: a review of eleven cases. Pediatr Dev Pathol 14 (2): 111-6, 2011 Mar-Apr.
- Stringer MD, Alizai NK: Mesenchymal hamartoma of the liver: a systematic review. J Pediatr Surg 40 (11): 1681-90, 2005.
- O'Sullivan MJ, Swanson PE, Knoll J, et al.: Undifferentiated embryonal sarcoma with unusual features arising within mesenchymal hamartoma of the liver: report of a case and review of the literature. Pediatr Dev Pathol 4 (5): 482-9, 2001 Sep-Oct.
- Walther A, Geller J, Coots A, et al.: Multimodal therapy including liver transplantation for hepatic undifferentiated embryonal sarcoma. Liver Transpl 20 (2): 191-9, 2014.
- Ismail H, Dembowska-Bagińska B, Broniszczak D, et al.: Treatment of undifferentiated embryonal sarcoma of the liver in children--single center experience. J Pediatr Surg 48 (11): 2202-6, 2013.
- Chowdhary SK, Trehan A, Das A, et al.: Undifferentiated embryonal sarcoma in children: beware of the solitary liver cyst. J Pediatr Surg 39 (1): E9-12, 2004.
- Baron PW, Majlessipour F, Bedros AA, et al.: Undifferentiated embryonal sarcoma of the liver successfully treated with chemotherapy and liver resection. J Gastrointest Surg 11 (1): 73-5, 2007.
- Kim DY, Kim KH, Jung SE, et al.: Undifferentiated (embryonal) sarcoma of the liver: combination treatment by surgery and chemotherapy. J Pediatr Surg 37 (10): 1419-23, 2002.
- Webber EM, Morrison KB, Pritchard SL, et al.: Undifferentiated embryonal sarcoma of the liver: results of clinical management in one center. J Pediatr Surg 34 (11): 1641-4, 1999.
- Bisogno G, Pilz T, Perilongo G, et al.: Undifferentiated sarcoma of the liver in childhood: a curable disease. Cancer 94 (1): 252-7, 2002.
- Urban CE, Mache CJ, Schwinger W, et al.: Undifferentiated (embryonal) sarcoma of the liver in childhood. Successful combined-modality therapy in four patients. Cancer 72 (8): 2511-6, 1993.
- Okajima H, Ohya Y, Lee KJ, et al.: Management of undifferentiated sarcoma of the liver including living donor liver transplantation as a backup procedure. J Pediatr Surg 44 (2): e33-8, 2009.
- Weitz J, Klimstra DS, Cymes K, et al.: Management of primary liver sarcomas. Cancer 109 (7): 1391-6, 2007.
- Plant AS, Busuttil RW, Rana A, et al.: A single-institution retrospective cases series of childhood undifferentiated embryonal liver sarcoma (UELS): success of combined therapy and the use of orthotopic liver transplant. J Pediatr Hematol Oncol 35 (6): 451-5, 2013.
- Mathias MD, Ambati SR, Chou AJ, et al.: A single-center experience with undifferentiated embryonal sarcoma of the liver. Pediatr Blood Cancer 63 (12): 2246-2248, 2016.
- Shi Y, Rojas Y, Zhang W, et al.: Characteristics and outcomes in children with undifferentiated embryonal sarcoma of the liver: A report from the National Cancer Database. Pediatr Blood Cancer 64 (4): , 2017.
- Techavichit P, Masand PM, Himes RW, et al.: Undifferentiated Embryonal Sarcoma of the Liver (UESL): A Single-Center Experience and Review of the Literature. J Pediatr Hematol Oncol 38 (4): 261-8, 2016.
- Merli L, Mussini C, Gabor F, et al.: Pitfalls in the surgical management of undifferentiated sarcoma of the liver and benefits of preoperative chemotherapy. Eur J Pediatr Surg 25 (1): 132-7, 2015.
- Kelly MJ, Martin L, Alonso M, et al.: Liver transplant for relapsed undifferentiated embryonal sarcoma in a young child. J Pediatr Surg 44 (12): e1-3, 2009.
Coriocarcinoma de hígado infantil (de lactantes)
El coriocarcinoma de hígado es un tumor muy raro que se origina en la placenta durante la gestación. Se presenta con una masa hepática en los primeros meses de vida. Debido a que en muchos casos hay metástasis desde la placenta a los tejidos maternos, se necesita hacer una prueba de gonadotropina coriónica humana ß (GCH-ß) a la madre. Con frecuencia, los lactantes están inestables en el momento del diagnóstico debido a hemorragia tumoral.
Diagnóstico
El diagnóstico clínico se puede hacer sin biopsia a partir de imágenes tumorales del hígado, concentraciones séricas excesivamente altas de GCH-ß y concentraciones normales de alfafetoproteína (AFP) para la edad.
Características histológicas
Se presentan citotrofoblastos y sincitiotrofoblastos. Los citotrofoblastos se configuran en nidos cohesionados de células de tamaño mediano con citoplasma claro, márgenes celulares característicos y núcleos vesiculares. Estos son sincitios multinucleados muy grandes formados a partir de los citotrofoblastos.
Pronóstico
El pronóstico de los lactantes con coriocarcinoma de hígado infantil a menudo es precario debido a la inestabilidad causada por la hemorragia en el momento de la presentación inicial. En un informe de caso y una revisión de la literatura de 32 informes de casos, se notificaron 6 sobrevivientes a largo plazo. Los autores han enfatizado en la importancia de un diagnóstico y tratamiento oportunos de este tumor que es muy quimiosensible.
Opciones de tratamiento del coriocarcinoma de hígado infantil (de lactantes)
Las opciones de tratamiento del coriocarcinoma de hígado infantil (de lactantes) son las siguientes:
- Resección quirúrgica.
- Quimioterapia seguida de resección quirúrgica.
- Quimioterapia seguida de trasplante de hígado.
Es posible que la extirpación quirúrgica inicial de la masa tumoral sea difícil por su friabilidad y tendencia hemorrágica. La extirpación quirúrgica del tumor primario a menudo se hace después de quimioterapia neoadyuvante.
Los tumores trofoblásticos gestacionales maternos son muy sensibles al metotrexato, y muchas mujeres, incluso aquellas con metástasis a distancia, se curan con monoquimioterapia. El coriocarcinoma materno y el infantil (de lactantes) se originan de la misma neoplasia maligna de placenta. La combinación de cisplatino, etopósido y bleomicina, tal como se usa para otros tumores de células germinativas infantiles, ha sido eficaz en algunos pacientes y a esta le sigue la resección de la masa residual. El uso de metotrexato neoadyuvante para el tratamiento del coriocarcinoma infantil (de lactantes), aunque a menudo produce una respuesta, no ha tenido un éxito uniforme.
En un informe de caso de quimioterapia neoadyuvante seguida de trasplante de hígado exitoso, se destaca la oportunidad de este tratamiento en niños cuyos tumores permanecen irresecables después de la quimioterapia.
Opciones de tratamiento en evaluación clínica para el coriocarcinoma de hígado infantil (de lactantes)
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
References
- Yoon JM, Burns RC, Malogolowkin MH, et al.: Treatment of infantile choriocarcinoma of the liver. Pediatr Blood Cancer 49 (1): 99-102, 2007.
- Olson T, Schneider D, Perlman E: Germ cell tumors. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 6th ed. Lippincott Williams and Wilkins, 2011, pp 1045-1067.
- Alsharif S, Karsou A: Infantile choriocarcinoma of the liver: case report and review of the literature. Oncol Cancer Case Rep 3 (1): 2017.
- Hanson D, Walter AW, Dunn S, et al.: Infantile choriocarcinoma in a neonate with massive liver involvement cured with chemotherapy and liver transplant. J Pediatr Hematol Oncol 33 (6): e258-60, 2011.
Tumores vasculares hepáticos
Para lograr un diagnóstico apropiado de los tumores vasculares hepáticos, es esencial prestar atención a los antecedentes clínicos, el examen físico, la evaluación de las pruebas de laboratorio y las imágenes radiológicas. Ante cualquier duda sobre la exactitud del diagnóstico, se debe realizar una biopsia.
El diagnóstico diferencial de los tumores vasculares hepáticos son los siguientes:
- Tumores benignos.
- Hemangiomas congénitos focales. Para obtener más información, consultar la sección Hemangiomas congénitos en Tratamiento de los tumores vasculares infantiles.
- Hemangiomas infantiles múltiples o difusos. Para obtener más información, consultar la sección Hemangioma infantil en Tratamiento de los tumores vasculares infantiles.
- Tumores malignos.
- Hemangioendotelioma epitelioide. Para obtener más información, consultar la sección Hemangioendotelioma epitelioide en Tratamiento de los tumores vasculares infantiles.
- Angiosarcoma. Para obtener más información, consultar la sección Angiosarcoma en Tratamiento de los tumores vasculares infantiles.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Actualizaciones más recientes a este resumen (03/15/2024)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Aspectos generales de las opciones de tratamiento del cáncer de hígado infantil
Se añadió a Shen et al. como referencia 16.
El Consejo editorial del PDQ sobre el tratamiento pediátrico es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este resumen del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento del cáncer de hígado infantil. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El Consejo editorial del PDQ sobre el tratamiento pediátrico, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
- Si el artículo se debe analizar en una reunión del consejo.
- Si conviene añadir texto acerca del artículo.
- Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Tratamiento del cáncer de hígado infantil son:
- Denise Adams, MD (Children's Hospital Boston)
- Andrea A. Hayes-Dixon, MD, FACS, FAAP (Howard University)
- Karen J. Marcus, MD, FACR (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Michael V. Ortiz, MD (Memorial Sloan Kettering Cancer Center)
- Stephen J. Shochat, MD (St. Jude Children's Research Hospital)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El Consejo editorial del PDQ sobre el tratamiento pediátrico emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como “En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]”.
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
PDQ® sobre el tratamiento pediátrico. PDQ Tratamiento del cáncer de hígado infantil. Bethesda, MD: National Cancer Institute. Actualización:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como “estándar” o “en evaluación clínica”. Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Comuníquese con el Instituto Nacional del Cáncer
Para obtener más información sobre las opciones para comunicarse con el NCI, incluso la dirección de correo electrónico, el número telefónico o el chat, consultar la página del Servicio de Información de Cáncer del Instituto Nacional del Cáncer.