Instituto Nacional del Cáncer
encontrar mi
Fecha de publicación: Jul 1, 2024
Resumen de información revisada por expertos acerca del tratamiento de la leucemia linfoblástica aguda infantil.
Tratamiento de la leucemia linfoblástica aguda infantil
Información general sobre la leucemia linfoblástica aguda infantil
El cáncer en niños y adolescentes es infrecuente, aunque desde 1975 se ha observado un aumento gradual de la incidencia general del cáncer infantil, incluso de la leucemia linfoblástica aguda (LLA). Se han logrado mejoras notables en la supervivencia de niños y adolescentes con cáncer. Entre 1975 y 2020, la mortalidad por cáncer infantil disminuyó en más del 50 %; sin embargo, el cáncer sigue siendo la causa principal de muerte por enfermedad después de la primera infancia entre los niños de los Estados Unidos. Para la LLA, la tasa de supervivencia a 5 años aumentó durante el mismo periodo desde el 60 % hasta casi el 90 % en los niños menores de 15 años, y desde el 28 % hasta más del 75 % en los adolescentes de 15 a 19 años. Los niños y adolescentes sobrevivientes de cáncer necesitan un seguimiento minucioso, ya que es posible que los efectos secundarios del tratamiento del cáncer persistan o se presenten meses o años después de este. Para obtener información específica sobre la incidencia, el tipo y la vigilancia de los efectos tardíos en los niños y adolescentes sobrevivientes de cáncer, consultar Efectos tardíos del tratamiento anticanceroso en la niñez.
Incidencia
La LLA es el cáncer que se diagnostica con mayor frecuencia en los niños, y representa alrededor del 25 % de los diagnósticos de cáncer en niños menores de 15 años. En los Estados Unidos, la tasa anual de la LLA es de casi 40 casos por millón de personas de 0 a 14 años de edad, y de casi 21 casos por millón de personas de 15 a 19 años. Cada año se diagnostica LLA a cerca de 3100 niños y adolescentes menores de 20 años en los Estados Unidos. Desde 1975, se ha producido un aumento gradual en la incidencia de la LLA.
Se observó un aumento marcado de la incidencia de LLA en los niños de 1 a 4 años (81 casos por millón al año); las tasas disminuyeron a 24 casos por millón a los 10 años de edad. La incidencia de la LLA en niños de 1 a 4 años de edad es cerca de 4 veces más alta que la incidencia en lactantes y niños de 10 años o más.
La incidencia de la LLA es más alta en los niños y adolescentes indígenas de las Américas o nativos de Alaska (65,9 casos por millón) y en los niños y adolescentes hispanos (48 casos por millón). La incidencia es mucho más alta en los niños blancos que en los niños negros, con una incidencia de LLA 2 veces más alta en niños blancos de 1 a 4 años que en los niños negros de la misma edad.
Características anatómicas
La LLA infantil se forma en los linfoblastos T y B en tejidos donde hay células progenitoras hematopoyéticas, como la médula ósea y el timo (consultar la Figura 1).
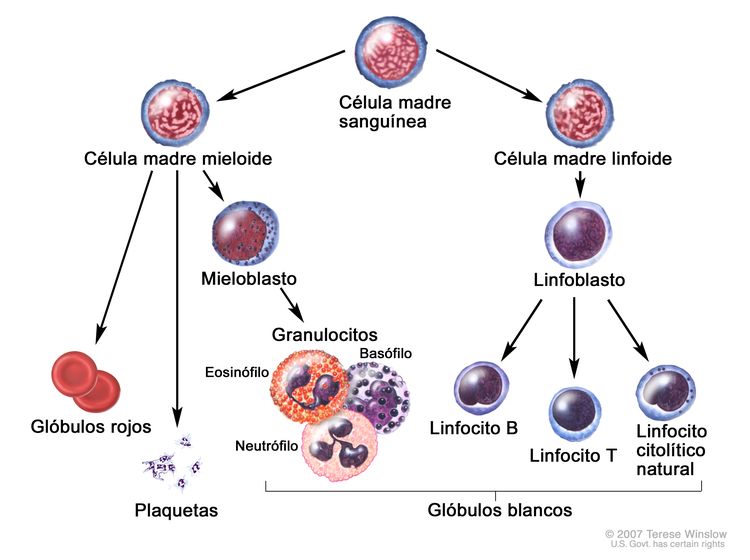 Figura 1. Evolución de una célula sanguínea. A partir de una misma célula madre se diferencian diversos linajes de células sanguíneas e inmunológicas, como los linfocitos T y los linfocitos B.
Figura 1. Evolución de una célula sanguínea. A partir de una misma célula madre se diferencian diversos linajes de células sanguíneas e inmunológicas, como los linfocitos T y los linfocitos B.
El compromiso medular de la leucemia aguda, según se observa en el microscopio óptico, se define de la siguiente manera.
- M1: menos de un 5 % de blastocitos.
- M2: de un 5 % a un 25 % de blastocitos.
- M3: más de un 25 % de blastocitos.
Casi todos los pacientes de LLA exhiben al inicio una médula de tipo M3.
Características morfológicas
En el pasado, los linfoblastos de la LLA se clasificaban según los criterios de la clasificación French-American-British (FAB) como tipo morfológico L1, L2 o L3. No obstante, este sistema de clasificación ya no se usa por la falta de importancia pronóstica independiente y su naturaleza subjetiva.
La mayoría de los casos de LLA que exhiben un tipo morfológico L3 expresan inmunoglobulina (lg) de superficie y tienen una translocación del gen MYC idéntica a la que se observa en el linfoma de Burkitt (es decir, t(8;14)(q24;q32), t(2;8)) que une el gen MYC con uno de los genes de la Ig. Los pacientes con esta forma poco frecuente de leucemia (leucemia de células B maduras o leucemia de Burkitt) se deben tratar de acuerdo con los protocolos para el linfoma de Burkitt. Para obtener más información acerca del tratamiento del linfoma o leucemia de células B maduras y del linfoma o leucemia de Burkitt, consultar Tratamiento del linfoma no Hodgkin infantil. Es infrecuente que los blastocitos de tipo morfológico L1/L2 (no L3) expresen Ig de superficie. Estos pacientes se deben tratar de la misma manera que los pacientes de LLA-B.
Factores de riesgo de la leucemia linfoblástica aguda
A continuación, se describen los principales factores de riesgo aceptados para la LLA y los genes asociados (cuando sea pertinente):
- Exposición prenatal a rayos X.
- Exposición posnatal a dosis altas de radiación (por ejemplo, radiación terapéutica que se usaba en el pasado para afecciones como la tiña de la cabeza o el crecimiento del timo).
- Tratamiento previo con quimioterapia.
- Afecciones genéticas como las siguientes:
- Síndrome de Down. Para obtener más información, consultar la sección Síndrome de Down.
- Neurofibromatosis (NF1).
- Síndrome de Bloom (BLM).
- Anemia de Fanconi (múltiples genes; la LLA es menos frecuente que la leucemia mieloide aguda [LMA]).
- Ataxia telangiectasia (ATM).
- Síndrome de Li-Fraumeni (TP53).
- Deficiencia constitucional de la reparación de errores de emparejamiento (variante bialélica de MLH1, MSH2, MSH6 y PMS2).
- Variantes genéticas hereditarias de penetrancia baja y alta. Para obtener más información, consultar la sección Variantes genéticas hereditarias de penetrancia baja y alta.
- Los portadores de una translocación robertsoniana constitucional que afecta los cromosomas 15 y 21 y los portadores de cromosoma 21 en anillo constitucional exhiben una predisposición específica muy alta a presentar una LLA con amplificación intracromosómica del cromosoma 21 (iAMP21).
Síndrome de Down
Los niños con síndrome de Down tienen un riesgo más alto de LLA y LMA; el riesgo acumulado de leucemia es de cerca del 2,1 % a los 5 años y del 2,7 % a los 30 años. Estas tasas representan un incremento del riesgo de LLA 20 a 30 veces mayor, y del riesgo de LMA más de 100 veces mayor, en los niños con síndrome de Down.
En un estudio de asociación de genoma completo se encontró que 4 locus de susceptibilidad a la LLA-B en una población sin síndrome de Down (IKZF1, CDKN2A, ARID5B y GATA3) también se vincularon con una susceptibilidad a la LLA en niños con síndrome de Down. La penetrancia del alelo de riesgo de CDKN2A fue más alta en los niños con síndrome de Down.
Entre la mitad y dos tercios de los casos de leucemia aguda en los niños con síndrome de Down son LLA y alrededor del 2 % al 3 % de los casos de LLA infantil se presentan en niños con este síndrome. La LLA en los niños con síndrome de Down exhibe una distribución por edad similar a la LLA en los niños sin síndrome de Down, con una mediana de edad de 3 a 4 años. Por el contrario, casi todos los casos de LMA en niños con síndrome de Down se presentan antes de los 4 años (mediana de edad, 1 año).
Los pacientes con LLA y síndrome de Down tienen una incidencia más baja de alteraciones genómicas favorables (fusión ETV6::RUNX1 e hiperdiploidía [51–65 cromosomas]), y desfavorables (fusiones BCR::ABL1 o KMT2A::AFF1 e hipodiploidía [<44 cromosomas]) así como un fenotipo de células T casi ausente.
Del 50 % al 60 % de los casos de LLA en niños con síndrome de Down exhiben alteraciones genómicas que afectan el gen CRLF2 y por lo general hay sobreexpresión de la proteína elaborada a partir de este gen, que establece un dímero con el receptor α de la interleucina-7 y conforma el receptor de la citocina linfopoyetina estromal tímica. En niños con síndrome de Down, sobre todo en aquellos de menor edad, la fusión P2RY8::CRLF2 se presenta con mucha más frecuencia que la fusión IGH::CRLF2. Se observan alteraciones genómicas de CRLF2 con una frecuencia mucho menor (<10 %) en los niños con LLA-B que no tienen síndrome de Down; cuando se presentan, por lo general se relacionan con el subtipo similar a BCR::ABL1. En un estudio retrospectivo, la frecuencia de reordenamientos de CRLF2 fue 9 veces más alta en niños con síndrome de Down y LLA que en niños con LLA, pero sin síndrome de Down (54,2 vs. 6,0 %). En ese estudio, solo el 25 % de los casos con reordenamientos de CRLF2 y síndrome de Down se clasificaron como similares a BCR::ABL1, en comparación con el 54 % de los casos con reordenamientos de CRLF2 sin síndrome de Down.
A partir de un número relativamente bajo de series publicadas, no se ha determinado la relevancia pronóstica de las alteraciones genómicas de CRLF2 en los pacientes con síndrome de Down y LLA. Sin embargo, entre los pacientes con síndrome de Down y reordenamientos de CRLF2, aquellos con la firma mutacional BCR::ABL1 presentan un pronóstico más precario que aquellos sin la fusión BCR::ABL1.
Alrededor del 20 % al 30 % de los casos de LLA que surgen en niños con síndrome de Down tienen variantes somáticas adquiridas en JAK2 o en JAK2, las cuales se relacionan de manera firme con reordenamientos de CRLF2. Lasvariantes de JAK son infrecuentes en niños más pequeños con LLA sin síndrome de Down, pero se observan con más frecuencia en niños de más edad y adolescentes con LLA-B de riesgo alto, sobre todo en aquellos con el subtipo similar a BCR::ABL1. La evidencia preliminar no indica ninguna correlación entre el estado de la variante de JAK2 y la supervivencia sin complicaciones (SSC) a 5 años en niños con síndrome de Down y LLA.
Las deleciones del gen IKZF1, que se observaron en el 20 % al 35 % de los pacientes con síndrome de Down y LLA, se relacionaron con un desenlace significativamente más precario en este grupo de pacientes.
Cerca del 10 % de los pacientes con síndrome de Down y LLA tienen alteraciones genómicas que producen la sobrexpresión o la activación anómala de los genes CEBPD, CEBPA y CEBPE. De los casos de LLA con activación de CEBP y síndrome de Down, alrededor del 40 % tienen también variantes puntuales o inserciones o deleciones de FLT3, en comparación con el 4,1% de los casos con síndrome de Down y otros subtipos de LLA.
Variantes genéticas hereditarias de penetrancia baja y alta
La predisposición genética a la LLA se divide en las siguientes categorías amplias:
- Predisposición asociada a síndromes genéticos. Aumento de riesgo de LLA asociado con los síndromes genéticos enumerados antes, aunque la LLA no sea la manifestación principal de la afección.
- Predisposición asociada a alelos comunes. Otra categoría de predisposición genética incluye los alelos comunes con efectos relativamente pequeños que se identifican mediante estudios de asociación de genoma completo. En estos estudios, se identificaron varios polimorfismos genéticos de la línea germinal (hereditarios) que se relacionan con la aparición de la LLA infantil. Por ejemplo, los alelos de riesgo de ARID5B se relacionan con la formación de LLA-B hiperdiploide (51–65 cromosomas). El gen ARID5B codifica un factor de transcripción importante para el desarrollo embrionario, la expresión génica específica por tipo celular y la regulación de la proliferación celular. Otros genes que exhiben polimorfismos relacionados con aumento del riesgo de LLA son GATA3, IKZF1, CDKN2A, CDKN2B, CEBPE, PIP4K2A y TP63.
Los factores de riesgo genético para la LLA-T se superponen con los factores de riesgo genético para la LLA-B, aunque algunos factores de riesgo son únicos. En un estudio de asociación de genoma completo se identificó un alelo de riesgo cerca de USP7 que se relacionó con aumento del riesgo de LLA-T (oportunidad relativa, 1,44) pero no LLA-B. El alelo de riesgo se relacionó con disminución en la transcripción de USP7, lo que es congruente con el hallazgo de variantes somáticas de pérdida de función de USP7 en pacientes con LLA-T. Las variantes germinales y somáticas de USP7 suelen ser mutuamente excluyentes y se observan con mayor frecuencia en pacientes de LLA-T con alteraciones de TAL1.
Los factores de riesgo genético que tienen un efecto similar en la aparición de la LLA-B y la LLA-T comprenden las alteraciones en CDKN2A, CDKN2B y 8q24.21 (variantes de la región amplificadora distal cis de MYC).
- Predisposición asociada a variantes de la línea germinal de penetrancia alta poco frecuentes. Las variantes de la línea germinal que producen cambios patógenos en genes relacionados con la LLA y que se observan en familias extensas con LLA familiar (es decir, tamaño grande del efecto) comprenden otra categoría de predisposición genética a la LLA. Muchos de los genes que se vinculan con el riesgo de LLA cumplen una función en el desarrollo de las células B (por ejemplo, PAX5, ETV6 y IKZF1).
- PAX5. En varias familias con múltiples casos de LLA, se identificó una variante de la línea germinal en PAX5 que cambia una glicina por serina en el aminoácido 183 y reduce la actividad de PAX5.
- ETV6. En familias extensas afectadas por trombocitopenia y LLA, se identificaron múltiples variantes de la línea germinal en ETV6 que producen la pérdida de la función de ETV6. En muestras de pacientes en remisión (es decir, en quienes se evalúa la línea germinal), la secuenciación de ETV6 permitió identificar variantes potencialmente relacionadas con la LLA en cerca del 1 % de los niños evaluados. Se observó que la mayoría de las variantes germinales (cerca del 75 %) eran deletéreas para el funcionamiento de ETV6, y el 70 % de los casos con una variante germinal deletérea en ETV6 tenían cariotipo hiperdiploide. El resto de los casos con una variante deletérea presentaban LLA diploide, con un perfil de transcripción similar al de los casos positivos para la fusión ETV6::RUNX1.
- TP53. Las variantes patógenas de la línea germinal en TP53 se asocian con un riesgo elevado de LLA. En un estudio de 3807 niños con LLA, se observó que 26 pacientes (0,7 %) tenían una variante patógena de la línea germinal en TP53 y una oportunidad relativa de 5,2 de presentar LLA. En comparación con la LLA en niños con TP53 natural o variantes de TP53 de significado incierto, la LLA en niños con variantes patógenas de la línea germinal en TP53 se asoció con mayor edad en el momento del diagnóstico (15,5 vs. 7,3 años), hipodiploidía (65 vs. 1 %), SSC y supervivencia general inferiores, así como un riesgo más alto de segundos cánceres.
- IKZF1. Las variantes de la línea germinal en IKZF1 se identificaron en una familia extensa con LLA familiar y en 43 de 4963 (0,9 %) niños con LLA esporádica. La mayoría (22 de 28) de las variantes de IKZF1 afectaron de manera adversa la función del gen IKZF1. Se identificaron variantes germinales en IKZF1 en la hipogammaglobulinemia hereditaria. En una serie, 2 de 29 pacientes presentaron LLA-B en la niñez.
Origen prenatal de la leucemia linfoblástica aguda infantil
En la mayoría de los casos, la leucemia linfoblástica aguda (LLA) se produce como consecuencia de un proceso de varios pasos que exige más de una alteración genómica para que se forme la leucemia evidente. La alteración genómica inicial en algunos casos de LLA infantil se produce en el útero. La evidencia que corrobora esto proviene de la detección en muestras sanguíneas de recién nacidos de reordenamientos de la inmunoglobulina o del antígeno del receptor de células T que son únicos en las células leucémicas de cada paciente. De modo similar, algunos pacientes con LLA caracterizada por anomalías cromosómicas específicas tienen glóbulos sanguíneos que exhiben por lo menos una anomalía genómica leucémica en el momento del nacimiento, y luego surgen otros cambios genómicos cooperativos adquiridos en el periodo posnatal. Los estudios genómicos en gemelos monocigóticos con leucemia coincidente respaldan aún más el origen prenatal de algunos tipos de leucemia.
También hay evidencia de que algunos niños que nunca presentan LLA nacen con unos glóbulos sanguíneos poco frecuentes, que presentan una alteración genómica relacionada con la LLA. Los estudios iniciales se enfocaron en la translocación ETV6::RUNX1, y en ellos se usó la reacción en cadena de la polimerasa (PCR) con retrotranscripción para identificar los transcritos de RNA que indican la presencia del gen de fusión. Por ejemplo, en un estudio, se encontró un resultado positivo para la translocación ETV6::RUNX1 en un 1 % de las gotas de sangre de recién nacidos (tarjetas de Guthrie). Aunque en informes posteriores en general se confirmó la presencia de la translocación ETV6::RUNX1 en algunos recién nacidos, las tasas y el grado de positividad variaron mucho.
Con el fin de abordar de manera definitiva esta pregunta, se usó una prueba de DNA con elevada sensibilidad y especificidad (PCR inversa genómica para la exploración de puntos de ruptura de ligadura) en 1000 muestras de sangre de cordón umbilical y se encontró la translocación ETV6::RUNX1 en un 5 % de las muestras. Cuando se aplicó el mismo método en 340 muestras de sangre de cordón umbilical, 2 muestras de cordón (0,6 %) fueron positivas para la fusión TCF3::PBX1. El porcentaje de muestras de sangre de cordón umbilical positivas para una de las translocaciones ETV6::RUNX1 o TCF3::PBX1 excede con creces el porcentaje de niños que presentará cualquier tipo de LLA (<0,01 %).
Cuadro clínico inicial
Se publicaron los síntomas típicos y atípicos, y los hallazgos clínicos de la LLA infantil.
Diagnóstico
Se publicó la evaluación necesaria para determinar el diagnóstico definitivo de la LLA infantil.
Pronóstico general
De los niños con LLA, alrededor del 98 % alcanzan la remisión, y se anticipa que cerca del 85 % de los pacientes de 1 a 18 años con LLA recién diagnosticada que reciben los regímenes actuales sobrevivirán sin complicaciones a largo plazo, y que más del 90 % seguirán vivos a los 5 años. En un estudio de pacientes con LLA de diagnóstico reciente, las recaídas fueron poco frecuentes (menos del 1 % de pacientes) al cabo de 6 a 7 años del diagnostico. Además, el exceso de riesgo de muerte asociado con el diagnóstico de leucemia ha disminuido de tal manera que la tasa de mortalidad de los sobrevivientes 6 a 7 años después del diagnóstico fue similar a la de la población general.
La presencia de hallazgos citogenéticos y genómicos, junto con los resultados de enfermedad residual mínima (ERM), permite definir subgrupos de LLA con tasas de SSC que superan el 95 %, y también lo contrario, es decir subgrupos con tasas de SSC del 50 % o menos. Para obtener más información, consultar las secciones Características citogenéticas y genómicas de la leucemia linfoblástica aguda infantil y Factores pronósticos que afectan el tratamiento según el riesgo.
A pesar de los avances en el tratamiento de la LLA infantil, todavía hay muchas dudas en el ámbito biológico y terapéutico que se deben resolver para lograr curar la LLA en todos los niños con la menor toxicidad posible. Para establecer una investigación sistemática de estas dudas se necesitan ensayos clínicos grandes y que se invite a participar a la mayoría de pacientes y familias.
Los ensayos clínicos para niños y adolescentes con LLA por lo general se diseñan para comparar el tratamiento que se acepta en la actualidad como estándar con regímenes en investigación que buscan mejorar las tasas de curación o disminuir la toxicidad. En ciertos ensayos en los que se obtiene una tasa de curación muy alta, se plantean preguntas sobre la reducción del tratamiento. La mayoría de los avances en la identificación de tratamientos curativos para la LLA infantil y otros cánceres infantiles se lograron gracias a los descubrimientos de investigación y las pruebas en ensayos clínicos controlados aleatorizados multicéntricos con diseño riguroso. Para obtener más información sobre ensayos clínicos en curso, consultar el portal de Internet del NCI.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
References
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed March 6, 2024.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 15, 2023.
- Childhood cancer. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 28. Also available online. Last accessed August 21, 2023.
- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 29. Also available online. Last accessed August 21, 2023.
- Howlader N, Noone AM, Krapcho M: SEER Cancer Statistics Review (CSR) 1975-2013. Bethesda, Md: National Cancer Institute, 2015. Available online. Last accessed June 04, 2021.
- Surveillance, Epidemiology, and End Results Program: SEER Cancer Stat Facts: Childhood Leukemia (Ages 0–19). Bethesda, Md: National Cancer Institute, DCCPS, Surveillance Research Program. Available online. Last accessed September 7, 2022.
- Special section: cancer in children and adolescents. In: American Cancer Society: Cancer Facts and Figures 2014. American Cancer Society, 2014, pp 25-42. Available online. Last accessed June 04, 2021.
- Shah A, Coleman MP: Increasing incidence of childhood leukaemia: a controversy re-examined. Br J Cancer 97 (7): 1009-12, 2007.
- Smith MA, Ries LA, Gurney JG, et al.: Leukemia. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 17-34. Also available online. Last accessed August 11, 2022.
- Barrington-Trimis JL, Cockburn M, Metayer C, et al.: Rising rates of acute lymphoblastic leukemia in Hispanic children: trends in incidence from 1992 to 2011. Blood 125 (19): 3033-4, 2015.
- Bennett JM, Catovsky D, Daniel MT, et al.: The morphological classification of acute lymphoblastic leukaemia: concordance among observers and clinical correlations. Br J Haematol 47 (4): 553-61, 1981.
- Koehler M, Behm FG, Shuster J, et al.: Transitional pre-B-cell acute lymphoblastic leukemia of childhood is associated with favorable prognostic clinical features and an excellent outcome: a Pediatric Oncology Group study. Leukemia 7 (12): 2064-8, 1993.
- Stiller CA, Chessells JM, Fitchett M: Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer 70 (5): 969-72, 1994.
- Passarge E: Bloom's syndrome: the German experience. Ann Genet 34 (3-4): 179-97, 1991.
- Alter BP: Cancer in Fanconi anemia, 1927-2001. Cancer 97 (2): 425-40, 2003.
- Taylor AM, Metcalfe JA, Thick J, et al.: Leukemia and lymphoma in ataxia telangiectasia. Blood 87 (2): 423-38, 1996.
- Holmfeldt L, Wei L, Diaz-Flores E, et al.: The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45 (3): 242-52, 2013.
- Powell BC, Jiang L, Muzny DM, et al.: Identification of TP53 as an acute lymphocytic leukemia susceptibility gene through exome sequencing. Pediatr Blood Cancer 60 (6): E1-3, 2013.
- Hof J, Krentz S, van Schewick C, et al.: Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol 29 (23): 3185-93, 2011.
- Ilencikova D, Sejnova D, Jindrova J, et al.: High-grade brain tumors in siblings with biallelic MSH6 mutations. Pediatr Blood Cancer 57 (6): 1067-70, 2011.
- Ripperger T, Schlegelberger B: Acute lymphoblastic leukemia and lymphoma in the context of constitutional mismatch repair deficiency syndrome. Eur J Med Genet 59 (3): 133-42, 2016.
- Moriyama T, Relling MV, Yang JJ: Inherited genetic variation in childhood acute lymphoblastic leukemia. Blood 125 (26): 3988-95, 2015.
- Li Y, Schwab C, Ryan SL, et al.: Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 508 (7494): 98-102, 2014.
- Harrison CJ, Moorman AV, Schwab C, et al.: An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia 28 (5): 1015-21, 2014.
- Hasle H: Pattern of malignant disorders in individuals with Down's syndrome. Lancet Oncol 2 (7): 429-36, 2001.
- Lupo PJ, Schraw JM, Desrosiers TA, et al.: Association Between Birth Defects and Cancer Risk Among Children and Adolescents in a Population-Based Assessment of 10 Million Live Births. JAMA Oncol 5 (8): 1150-1158, 2019.
- Marlow EC, Ducore J, Kwan ML, et al.: Leukemia Risk in a Cohort of 3.9 Million Children with and without Down Syndrome. J Pediatr 234: 172-180.e3, 2021.
- Brown AL, de Smith AJ, Gant VU, et al.: Inherited genetic susceptibility to acute lymphoblastic leukemia in Down syndrome. Blood 134 (15): 1227-1237, 2019.
- Zeller B, Gustafsson G, Forestier E, et al.: Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br J Haematol 128 (6): 797-804, 2005.
- Arico M, Ziino O, Valsecchi MG, et al.: Acute lymphoblastic leukemia and Down syndrome: presenting features and treatment outcome in the experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP). Cancer 113 (3): 515-21, 2008.
- Maloney KW, Carroll WL, Carroll AJ, et al.: Down syndrome childhood acute lymphoblastic leukemia has a unique spectrum of sentinel cytogenetic lesions that influences treatment outcome: a report from the Children's Oncology Group. Blood 116 (7): 1045-50, 2010.
- de Graaf G, Buckley F, Skotko BG: Estimation of the number of people with Down syndrome in the United States. Genet Med 19 (4): 439-447, 2017.
- Chessells JM, Harrison G, Richards SM, et al.: Down's syndrome and acute lymphoblastic leukaemia: clinical features and response to treatment. Arch Dis Child 85 (4): 321-5, 2001.
- Buitenkamp TD, Izraeli S, Zimmermann M, et al.: Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood 123 (1): 70-7, 2014.
- Hertzberg L, Vendramini E, Ganmore I, et al.: Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood 115 (5): 1006-17, 2010.
- Buitenkamp TD, Pieters R, Gallimore NE, et al.: Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia 26 (10): 2204-11, 2012.
- Mullighan CG, Collins-Underwood JR, Phillips LA, et al.: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243-6, 2009.
- Russell LJ, Jones L, Enshaei A, et al.: Characterisation of the genomic landscape of CRLF2-rearranged acute lymphoblastic leukemia. Genes Chromosomes Cancer 56 (5): 363-372, 2017.
- Harvey RC, Mullighan CG, Chen IM, et al.: Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 115 (26): 5312-21, 2010.
- Schwab CJ, Chilton L, Morrison H, et al.: Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica 98 (7): 1081-8, 2013.
- Li Z, Chang TC, Junco JJ, et al.: Genomic landscape of Down syndrome-associated acute lymphoblastic leukemia. Blood 142 (2): 172-184, 2023.
- Bercovich D, Ganmore I, Scott LM, et al.: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484-92, 2008.
- Gaikwad A, Rye CL, Devidas M, et al.: Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol 144 (6): 930-2, 2009.
- Kearney L, Gonzalez De Castro D, Yeung J, et al.: Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood 113 (3): 646-8, 2009.
- Mullighan CG, Zhang J, Harvey RC, et al.: JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 106 (23): 9414-8, 2009.
- Hanada I, Terui K, Ikeda F, et al.: Gene alterations involving the CRLF2-JAK pathway and recurrent gene deletions in Down syndrome-associated acute lymphoblastic leukemia in Japan. Genes Chromosomes Cancer 53 (11): 902-10, 2014.
- Michels N, Boer JM, Enshaei A, et al.: Minimal residual disease, long-term outcome, and IKZF1 deletions in children and adolescents with Down syndrome and acute lymphocytic leukaemia: a matched cohort study. Lancet Haematol 8 (10): e700-e710, 2021.
- Papaemmanuil E, Hosking FJ, Vijayakrishnan J, et al.: Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet 41 (9): 1006-10, 2009.
- Treviño LR, Yang W, French D, et al.: Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet 41 (9): 1001-5, 2009.
- Migliorini G, Fiege B, Hosking FJ, et al.: Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood 122 (19): 3298-307, 2013.
- Hungate EA, Vora SR, Gamazon ER, et al.: A variant at 9p21.3 functionally implicates CDKN2B in paediatric B-cell precursor acute lymphoblastic leukaemia aetiology. Nat Commun 7: 10635, 2016.
- Sherborne AL, Hosking FJ, Prasad RB, et al.: Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet 42 (6): 492-4, 2010.
- Xu H, Yang W, Perez-Andreu V, et al.: Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst 105 (10): 733-42, 2013.
- Ellinghaus E, Stanulla M, Richter G, et al.: Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia 26 (5): 902-9, 2012.
- Qian M, Zhao X, Devidas M, et al.: Genome-Wide Association Study of Susceptibility Loci for T-Cell Acute Lymphoblastic Leukemia in Children. J Natl Cancer Inst 111 (12): 1350-1357, 2019.
- Somasundaram R, Prasad MA, Ungerbäck J, et al.: Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood 126 (2): 144-52, 2015.
- Shah S, Schrader KA, Waanders E, et al.: A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet 45 (10): 1226-31, 2013.
- Auer F, Rüschendorf F, Gombert M, et al.: Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia 28 (5): 1136-8, 2014.
- Zhang MY, Churpek JE, Keel SB, et al.: Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet 47 (2): 180-5, 2015.
- Topka S, Vijai J, Walsh MF, et al.: Germline ETV6 Mutations Confer Susceptibility to Acute Lymphoblastic Leukemia and Thrombocytopenia. PLoS Genet 11 (6): e1005262, 2015.
- Noetzli L, Lo RW, Lee-Sherick AB, et al.: Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet 47 (5): 535-8, 2015.
- Rampersaud E, Ziegler DS, Iacobucci I, et al.: Germline deletion of ETV6 in familial acute lymphoblastic leukemia. Blood Adv 3 (7): 1039-1046, 2019.
- Nishii R, Baskin-Doerfler R, Yang W, et al.: Molecular basis of ETV6-mediated predisposition to childhood acute lymphoblastic leukemia. Blood 137 (3): 364-373, 2021.
- Qian M, Cao X, Devidas M, et al.: TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J Clin Oncol 36 (6): 591-599, 2018.
- Churchman ML, Qian M, Te Kronnie G, et al.: Germline Genetic IKZF1 Variation and Predisposition to Childhood Acute Lymphoblastic Leukemia. Cancer Cell 33 (5): 937-948.e8, 2018.
- Kuehn HS, Boisson B, Cunningham-Rundles C, et al.: Loss of B Cells in Patients with Heterozygous Mutations in IKAROS. N Engl J Med 374 (11): 1032-1043, 2016.
- Greaves MF, Wiemels J: Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer 3 (9): 639-49, 2003.
- Taub JW, Konrad MA, Ge Y, et al.: High frequency of leukemic clones in newborn screening blood samples of children with B-precursor acute lymphoblastic leukemia. Blood 99 (8): 2992-6, 2002.
- Bateman CM, Colman SM, Chaplin T, et al.: Acquisition of genome-wide copy number alterations in monozygotic twins with acute lymphoblastic leukemia. Blood 115 (17): 3553-8, 2010.
- Greaves MF, Maia AT, Wiemels JL, et al.: Leukemia in twins: lessons in natural history. Blood 102 (7): 2321-33, 2003.
- Mori H, Colman SM, Xiao Z, et al.: Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A 99 (12): 8242-7, 2002.
- Schäfer D, Olsen M, Lähnemann D, et al.: Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood 131 (7): 821-826, 2018.
- Hein D, Dreisig K, Metzler M, et al.: The preleukemic TCF3-PBX1 gene fusion can be generated in utero and is present in ≈0.6% of healthy newborns. Blood 134 (16): 1355-1358, 2019.
- Gramatges MM, O'Brien MM, Rabin KR: Acute lymphoblastic leukemia. In: Blaney SM, Helman LJ, Adamson PC, eds.: Pizzo and Poplack's Pediatric Oncology. 8th ed. Wolters Kluwer, 2020, pp 419-53.
- Chessells JM; haemostasis and thrombosis task force, British committee for standards in haematology: Pitfalls in the diagnosis of childhood leukaemia. Br J Haematol 114 (3): 506-11, 2001.
- Onciu M: Acute lymphoblastic leukemia. Hematol Oncol Clin North Am 23 (4): 655-74, 2009.
- Margolskee E, Waith Wertheim GB, Harvey RC: Pathology and molecular diagnosis of leukemias and lymphomas. In: Blaney SM, Helman LJ, Adamson PC, eds.: Pizzo and Poplack's Pediatric Oncology. 8th ed. Wolters Kluwer, 2020, pp 117-30.
- Cheng J, Klairmont MM, Choi JK: Peripheral blood flow cytometry for the diagnosis of pediatric acute leukemia: Highly reliable with rare exceptions. Pediatr Blood Cancer 66 (1): e27453, 2019.
- Möricke A, Zimmermann M, Valsecchi MG, et al.: Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood 127 (17): 2101-12, 2016.
- Vora A, Goulden N, Wade R, et al.: Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol 14 (3): 199-209, 2013.
- Place AE, Stevenson KE, Vrooman LM, et al.: Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): a randomised, open-label phase 3 trial. Lancet Oncol 16 (16): 1677-90, 2015.
- Pieters R, de Groot-Kruseman H, Van der Velden V, et al.: Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol 34 (22): 2591-601, 2016.
- Moorman AV, Antony G, Wade R, et al.: Time to Cure for Childhood and Young Adult Acute Lymphoblastic Leukemia Is Independent of Early Risk Factors: Long-Term Follow-Up of the UKALL2003 Trial. J Clin Oncol 40 (36): 4228-4239, 2022.
Sistema de clasificación de la Organización Mundial de la Salud para la leucemia linfoblástica aguda infantil
En la 5ª edición de la clasificación de tumores de tejidos hematopoyéticos y linfoides de la Organización Mundial de la Salud (OMS), se enumeran las siguientes entidades relacionadas con las leucemias linfoides agudas:
5ª edición de la clasificación de leucemias o linfomas linfoblásticos de células B de la OMS
- Leucemia o linfoma linfoblásticos de células B, sin otra indicación (SAI).
- Leucemia o linfoma linfoblásticos de células B con hiperdiploidía alta.
- Leucemia o linfoma linfoblásticos de células B con iAMP21.
- Leucemia o linfoma linfoblásticos de células B con la fusión BCR::ABL1.
- Leucemia o linfoma linfoblásticos de células B con características similares a BCR::ABL1.
- Leucemia o linfoma linfoblásticos de células B con reordenamiento de KMT2A.
- Leucemia o linfoma linfoblásticos de células B con la fusión ETV6::RUNX1.
- Leucemia o linfoma linfoblásticos de células B con características similares a ETV6::RUNX1.
- Leucemia o linfoma linfoblásticos de células B con la fusión TCF3::PBX1.
- Leucemia o linfoma linfoblásticos de células B con la fusión IGH::ABL1.
- Leucemia o linfoma linfoblásticos de células B con la fusión TCF3::HLF.
- Leucemia o linfoma linfoblásticos de células B con otras anomalías genéticas caracterizadas.
La categoría de LLA-B con otras anomalías genéticas caracterizadas incluye posibles entidades nuevas, como la LLA-B con reordenamientos de DUX4, MEF2D, ZNF384 o NUTM1; la LLA-B con fusiones IG::MYC; y la LLA-B con anomalías PAX5alt o PAX5 p.P80R (NP_057953.1).
5ª edición de la clasificación de leucemias o linfomas linfoblásticos de células T de la OMS
- Leucemia o linfoma linfoblásticos de células T, SAI.
- Leucemia o linfoma linfoblásticos de células T precursoras tempranas.
Clasificación de la OMS de 2016 para las leucemias agudas de linaje ambiguo
En el Cuadro 1 se resume el sistema de clasificación de la OMS para el grupo de leucemias agudas de linaje ambiguo, es decir, que exhiben características de leucemia mieloide aguda (LMA) y leucemia linfoblástica aguda (LLA) 1. Los criterios de asignación de linaje para el diagnóstico de la leucemia aguda de fenotipo mixto (LAFM) se presentan en el Cuadro 2.
| Afección | Definición |
|---|---|
| LAFM = leucemia aguda de fenotipo mixto; SAI = sin otra indicación. | |
| aAdaptación de Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009. Del portal de Internet del Haematologica/the Hematology Journal (http://www.haematologica.org). | |
| Leucemia aguda indiferenciada | Leucemia aguda que no expresa ningún marcador que se considere específico para el linaje linfoide ni el linaje mieloide |
| LAFM con BCR::ABL1 (t(9;22)(q34;q11.2)) | Leucemia aguda que cumple con los criterios diagnósticos de la leucemia aguda de fenotipo mixto en la que los blastocitos también expresan la translocación (9;22) o el reordenamiento BCR::ABL1 |
| LAFM con el reordenamiento KMT2A (t(v;11q23)) | Leucemia aguda que cumple con los criterios diagnósticos de la leucemia aguda de fenotipo mixto en la que los blastocitos también expresan una translocación que afecta el gen KMT2A |
| LAFM, B/mieloide, SAI (LAFM B/M) | Leucemia aguda que cumple con los criterios diagnósticos para que se le asigne un linaje B y un linaje mieloide, en la que los blastocitos carecen de anomalías genéticas que afectan BCR::ABL1 o KMT2A |
| LAFM, T/mieloide, SAI (LAFM T/M) | Leucemia aguda que cumple con los criterios diagnósticos para que se le asigne un linaje T y un linaje mieloide, en la que los blastocitos carecen de anomalías genéticas que afectan BCR::ABL1 o KMT2A |
| LAFM, B/mieloide, SAI—tipos poco comunes | Leucemia aguda que cumple con los criterios diagnósticos para que se le asigne un linaje B y un linaje T |
| Otras leucemias de linaje ambiguo | Leucemia o linfoma linfoblásticos de células citolíticas naturales |
| Linaje | Criterios |
|---|---|
| aAdaptado de Arber et al. | |
| bFuerte se define como igual o más brillante que las células B o T normales en la muestra. | |
| Linaje mieloide | Mieloperoxidasa (citometría de flujo, prueba inmunohistoquímica o citoquímica); o diferenciación monocítica (por lo menos dos de los siguientes aspectos: prueba citoquímica de esterasa inespecífica, CD11c, CD14, CD64 o lisozima) |
| Linaje T | Fuerteb CD3 citoplasmático (con anticuerpos contra la cadena ε de CD3); o CD3 de superficie |
| Linaje B | Fuerteb CD19 fuerte y expresión fuerte de por lo menos una de las siguientes moléculas: CD79a, CD22 citoplasmático o CD10; o CD19 débil y expresión fuerte de por lo menos dos de las siguientes moléculas: CD79a, CD22 citoplasmático o CD10 |
Es posible que se observen leucemias de fenotipos mixtos en distintas presentaciones; por ejemplo, las siguientes:
- Leucemias bilineales en las que hay dos poblaciones de células diferentes; a menudo, una linfoide y otra mieloide.
- Leucemias bifenotípicas en las que los blastocitos exhiben a la vez características de linaje linfoide y mieloide.
Los casos bifenotípicos representan la mayoría de las leucemias de fenotipo mixto. Los pacientes con leucemias bifenotípicas de linaje B y mieloide que carecen de la fusión ETV6::RUNX1 tienen tasas más bajas de remisión completa (RC) y una supervivencia sin complicaciones (SSC) significativamente más precaria, en comparación con los pacientes de LLA-B. Se han notificado casos de LAFM (B/mieloide) que exhiben fusiones génicas de ZNF384, y en la evaluación genómica de la LAFM se encontró que las fusiones génicas de ZNF384 estaban presentes en cerca de la mitad de los casos con fenotipo B/mieloide.
En algunos estudios se indica que los pacientes con leucemia bifenotípica a veces evolucionan mejor con un régimen de tratamiento linfoide que con uno mieloide.; [Nivel de evidencia C1] En un estudio retrospectivo grande del grupo internacional Berlin-Frankfurt-Münster se demostró que el tratamiento inicial con un régimen de tipo LLA se relacionó con un desenlace superior al régimen de tipo LMA o los regímenes combinados para LLA y LMA; en particular, en los casos positivos para CD19 o con otra expresión de antígenos linfoides. En este estudio, el trasplante de células madre hematopoyéticas durante la primera RC no fue beneficioso, con la posible excepción de casos con hallazgos morfológicos de enfermedad persistente en la médula ósea (≥5 % de blastocitos) después del primer mes de tratamiento.
Para obtener más información sobre características clínicas y biológicas esenciales, así como la importancia pronóstica de estas entidades, consulte la sección Características citogenéticas y genómicas de la leucemia linfoblástica aguda infantil.
References
- Alaggio R, Amador C, Anagnostopoulos I, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 36 (7): 1720-1748, 2022.
- Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.
- Borowitz MJ, Béné MC, Harris NL: Acute leukaemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 150-5.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Gerr H, Zimmermann M, Schrappe M, et al.: Acute leukaemias of ambiguous lineage in children: characterization, prognosis and therapy recommendations. Br J Haematol 149 (1): 84-92, 2010.
- Shago M, Abla O, Hitzler J, et al.: Frequency and outcome of pediatric acute lymphoblastic leukemia with ZNF384 gene rearrangements including a novel translocation resulting in an ARID1B/ZNF384 gene fusion. Pediatr Blood Cancer 63 (11): 1915-21, 2016.
- Yao L, Cen J, Pan J, et al.: TAF15-ZNF384 fusion gene in childhood mixed phenotype acute leukemia. Cancer Genet 211: 1-4, 2017.
- Alexander TB, Gu Z, Iacobucci I, et al.: The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 562 (7727): 373-379, 2018.
- Rubnitz JE, Onciu M, Pounds S, et al.: Acute mixed lineage leukemia in children: the experience of St Jude Children's Research Hospital. Blood 113 (21): 5083-9, 2009.
- Al-Seraihy AS, Owaidah TM, Ayas M, et al.: Clinical characteristics and outcome of children with biphenotypic acute leukemia. Haematologica 94 (12): 1682-90, 2009.
- Matutes E, Pickl WF, Van't Veer M, et al.: Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood 117 (11): 3163-71, 2011.
- Hrusak O, de Haas V, Stancikova J, et al.: International cooperative study identifies treatment strategy in childhood ambiguous lineage leukemia. Blood 132 (3): 264-276, 2018.
- Orgel E, Alexander TB, Wood BL, et al.: Mixed-phenotype acute leukemia: A cohort and consensus research strategy from the Children's Oncology Group Acute Leukemia of Ambiguous Lineage Task Force. Cancer 126 (3): 593-601, 2020.
Características citogenéticas y genómicas de la leucemia linfoblástica aguda infantil
Características genómicas de la leucemia linfoblástica aguda infantil
Se han investigado a fondo las características genómicas de la leucemia linfoblástica aguda (LLA) infantil y se definieron múltiples subtipos diferenciados a partir de la caracterización citogenética y molecular; cada subtipo con su propio perfil de características clínicas y pronósticas. El análisis de las características genómicas de la LLA infantil se divide a continuación en 3 secciones: las alteraciones genómicas de la LLA-B, de la LLA-T y de la leucemia aguda de fenotipo mixto (LAFM). En las Figuras 2, 3, y 5 se observa la distribución de los casos de LLA-B (estratificados de acuerdo a la LLA-B de riesgo estándar y de riesgo alto del Instituto Nacional del Cáncer [NCI]) y LLA-T según los subtipos citogenético y molecular.
En esta sección, los porcentajes de los subtipos genómicos entre todos los casos de LLA-B y LLA-T provienen, sobre todo, de un informe que describe la caracterización genómica de pacientes tratados en varios ensayos clínicos del Children's Oncology Group (COG) y el St. Jude Children's Research Hospital (SJCRH). Se presentan los porcentajes por subtipo para los pacientes con LLA-B de riesgo estándar y riesgo alto según el NCI (hasta los 18 años).
Características citogenéticas y genómicas de la leucemia linfoblástica aguda de células B
La LLA-B se clasifica de acuerdo a las siguientes alteraciones genómicas: 1) fusiones génicas que producen factores de transcripción con actividad anómala, 2) ganancias y pérdidas cromosómicas (por ejemplo, hiperdiploidía o hipodiploidía) y 3) alteraciones que producen la activación de genes de tirosina–cinasas. En las Figuras 2 y 3 se muestra la distribución de los casos de LLA-B de riesgo estándar y riesgo alto del NCI en 23 subtipos citogenéticos y moleculares. Los 2 subtipos más comunes (hiperdiploide y de fusión ETV6::RUNX1) representan juntos alrededor del 60 % de los casos de LLA-B de riesgo estándar del NCI, pero solo cerca del 25 % de los casos de riesgo alto del NCI. La mayoría de los otros subtipos son mucho menos frecuentes, y representan menos del 2 % al 3 % de los casos de LLA-B. Las características moleculares y clínicas de algunos de los subtipos se analizan más adelante.
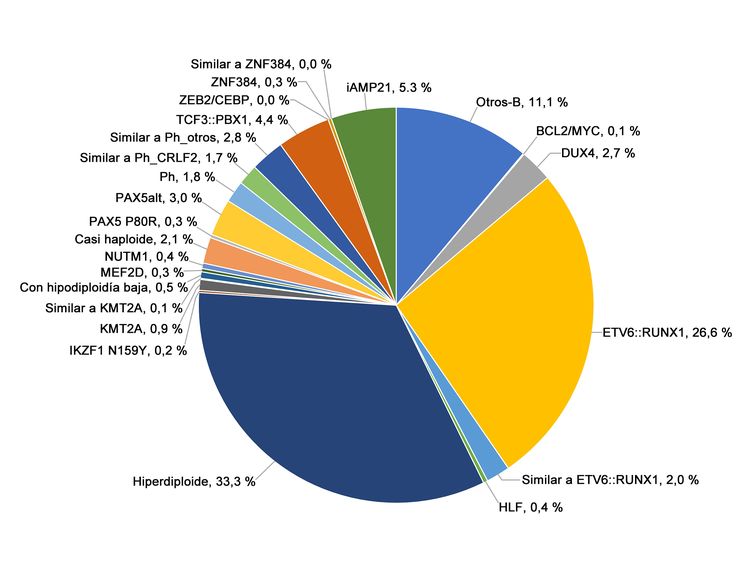 Figura 2. Frecuencia de los subtipos genómicos de la LLA-B de riesgo estándar del NCI. En la figura se observan los datos de 1126 niños diagnosticados con LLA-B de riesgo estándar del NCI (edad, 1–9 años y RGB <50 000/µL) e inscritos en los ensayos clínicos del St. Jude Children’s Research Hospital o el Children’s Oncology Group. Adaptado del cuadro 2 suplementario de Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
Figura 2. Frecuencia de los subtipos genómicos de la LLA-B de riesgo estándar del NCI. En la figura se observan los datos de 1126 niños diagnosticados con LLA-B de riesgo estándar del NCI (edad, 1–9 años y RGB <50 000/µL) e inscritos en los ensayos clínicos del St. Jude Children’s Research Hospital o el Children’s Oncology Group. Adaptado del cuadro 2 suplementario de Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
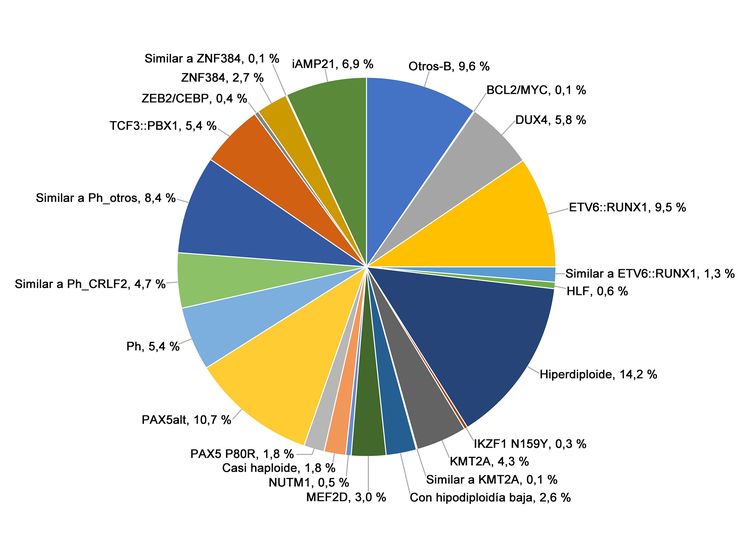 Figura 3. Frecuencia de los subtipos genómicos de la LLA-B de riesgo alto del NCI. En la figura se observan los datos de 1084 niños diagnosticados con LLA-B de riesgo alto del NCI (edad, 1–18 años y RGB > 50 000/µL) e inscritos en los ensayos clínicos del St. Jude Children’s Research Hospital o el Children’s Oncology Group. Adaptado del cuadro 2 suplementario de Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
Figura 3. Frecuencia de los subtipos genómicos de la LLA-B de riesgo alto del NCI. En la figura se observan los datos de 1084 niños diagnosticados con LLA-B de riesgo alto del NCI (edad, 1–18 años y RGB > 50 000/µL) e inscritos en los ensayos clínicos del St. Jude Children’s Research Hospital o el Children’s Oncology Group. Adaptado del cuadro 2 suplementario de Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
El panorama genómico de la LLA-B se caracteriza por una serie de alteraciones genómicas que interrumpen el desarrollo normal de las células B y, en algunos casos, por variantes de genes que proporcionan una señal de proliferación (por ejemplo, variantes activadoras de los genes de la familia RAS o variantes y translocaciones que producen señalización mediante una vía de cinasa). Las alteraciones genómicas que interrumpen el desarrollo de las células B son, entre otras, translocaciones (por ejemplo, las fusiones TCF3::PBX1 y ETV6::RUNX1), variantes puntuales (por ejemplo, de IKZF1 y PAX5), y deleciones intragénicas o intergénicas (por ejemplo, de IKZF1, PAX5, EBF y ERG).
Las alteraciones genómicas de la LLA-B no suelen ocurrir al azar, más bien se agrupan en los subtipos demarcados por sus características biológicas y perfiles de expresión génica. Los casos con translocaciones cromosómicas recurrentes (por ejemplo, las fusiones TCF3::PBX1 y ETV6::RUNX1, y la LLA con reordenamiento de KMT2A) exhiben características biológicas distintivas que ilustran este punto, al igual que los siguientes ejemplos de alteraciones genómicas específicas dentro de subtipos biológicos únicos:
- Las deleciones y variantes de IKZF1 se observan con mayor frecuencia en los casos de LLA con BCR::ABL1 y LLA similar a BCR::ABL1.
- Las deleciones intragénicas de ERG se presentan en un subtipo diferenciado caracterizado por reordenamientos del gen DUX4.
- Las variantes de TP53, a menudo de la línea germinal, son muy frecuentes en pacientes de LLA con hipodiploidía baja de 32 a 39 cromosomas. Las variantes de TP53 son infrecuentes en otros pacientes de LLA-B.
Las variantes puntuales activadoras de genes de cinasas son infrecuentes en la LLA-B de riesgo alto. Los genes de las cinasas JAK son los genes de cinasas que se encuentran alterados con mayor frecuencia. Por lo general, estas variantes se observan en los pacientes con LLA similar a BCR::ABL1 que tienen anomalías en CRLF2, aunque también se observan variantes de JAK2 en cerca del 25 % de los niños con síndrome de Down y LLA, que se presenta solo en casos con reordenamientos génicos de CRLF2. Varios genes de cinasas y receptores de citocinas se activan mediante translocaciones, como se describe a continuación en el análisis de la LLA con BCR::ABL1 y la LLA similar a BCR::ABL1. Se presentan variantes de FLT3 en una minoría de los casos (alrededor del 10 %) de LLA hiperdiploide y LLA con reordenamiento de KMT2A; estas variantes son escasas en otros subtipos.
La comprensión de las características genómicas de la LLA-B en el momento de la recaída está menos avanzada que la comprensión de las características genómicas de la LLA en el momento del diagnóstico. A menudo, la LLA infantil es policlonal en el momento del diagnóstico y, por influencia selectiva del tratamiento, algunos clones se extinguen mientras que surgen clones nuevos con perfiles genómicos diferenciados. Sin embargo, el subtipo molecular que define lesiones como translocaciones y aneuploidía casi siempre se mantiene en el momento de la recaída. Las variantes nuevas que surgen en el momento de la recaída son de particular importancia porque su selección quizás se produzca por efecto de componentes específicos del tratamiento. Por ejemplo, en dos estudios de pacientes con LLA-B no se encontraron variantes de NT5C2 en el momento del diagnóstico, pero durante la recaída temprana se observaron variantes específicas de NT5C2 en 7 de 44 pacientes (16 %) y 9 de 20 (45 %) pacientes de cada estudio. Las variantes de NT5C2 son poco frecuentes en los pacientes con una recaída tardía, estas variantes inducen resistencia a la mercaptopurina y a la tioguanina. Otro gen que se encuentra alterado solamente en el momento de la recaída es PRSP1, un gen que participa en la biosíntesis de las purinas. Se observaron variantes en el 13,0 % de los pacientes de una cohorte china y en el 2,7 % de los pacientes de una cohorte alemana; estas se observaron en pacientes con recaídas durante el tratamiento. Las variantes de PRSP1 observadas en los casos de recaída inducen resistencia a las tiopurinas en líneas celulares de leucemia. Las variantes de CREBBP también son muy frecuentes en el momento de la recaída y se vinculan con una resistencia elevada a los glucocorticoides. Es posible que una mayor comprensión de las características genómicas en el momento de la recaída permita adaptar el tratamiento inicial para evitar las recaídas o detectar de manera temprana las variantes que producen resistencia, de manera que se logre una intervención antes de que se produzca una recaída evidente.
Se ha observado que varias anomalías cromosómicas recurrentes tienen importancia pronóstica; en especial, para la LLA-B. Algunas anomalías cromosómicas se relacionan con desenlaces más favorables, como las trisomías de pronóstico favorable (51–65 cromosomas) y la fusión ETV6::RUNX1.[Nivel de evidencia B4] Otras alteraciones como la fusión BCR::ABL1 (que genera el resultado positivo para el cromosoma Filadelfia [Ph+]; t(9;22)(q34;q11.2)), los reordenamientos en el gen KMT2A, la hipodiploidía y la amplificación intracromosómica del gen RUNX1 (iAMP21), tradicionalmente se han relacionado con un desenlace más precario.
En reconocimiento de la importancia clínica de muchas de estas alteraciones genómicas, en la quinta edición de la revisión de la Classification of Haematolymphoid Tumours se enumeran las siguientes entidades para la LLA-B:
- Leucemia o linfoma linfoblástico-B, sin otra indicación (SAI).
- Leucemia o linfoma linfoblástico-B con hiperdiploidía alta.
- Leucemia o linfoma linfoblástico-B con hipodiploidía.
- Leucemia o linfoma linfoblástico-B con iAMP21.
- Leucemia o linfoma linfoblástico-B con la fusión BCR::ABL1.
- Leucemia o linfoma linfoblástico-B con características similares a BCR::ABL1.
- Leucemia o linfoma linfoblástico-B con reordenamientos en KMT2A.
- Leucemia o linfoma linfoblástico-B con la fusión ETV6::RUNX1.
- Leucemia o linfoma linfoblástico-B con características similares a ETV6::RUNX1.
- Leucemia o linfoma linfoblástico-B con la fusión TCF3::PBX1.
- Leucemia o linfoma linfoblástico-B con la fusión IGH::IL3.
- Leucemia o linfoma linfoblástico-B con la fusión TCF3::HLF.
- Leucemia o linfoma linfoblástico-B con otras anomalías genéticas definidas.
La categoría de LLA-B con otras anomalías genéticas definidas incluye posibles entidades nuevas, como LLA-B con reordenamientos en DUX4, MEF2D, ZNF384 o NUTM1; LLA-B con fusiones IG::MYC; y LLA-B con anomalías en PAX5alt o PAX5 p.P80R (NP_057953.1).
Estas y otras anomalías cromosómicas y genómicas de la LLA infantil se describen a continuación.
- Número de cromosomas.
- Hiperdiploidía alta (51–65 cromosomas).
En los pacientes pediátricos con LLA-B, la hiperdiploidía alta (presencia de 51 a 65 cromosomas por célula o un índice de DNA superior a 1,16), se presenta en alrededor del 33 % de los casos de riesgo estándar del NCI y del 14 % de los casos de riesgo alto del NCI. Es posible evaluar la hiperdiploidía por la medición del contenido de DNA en las células (índice de DNA) o por cariotipado. En los casos que exhiben un cariotipo normal o en los que el análisis citogenético estándar fue insatisfactorio, la hibridación fluorescente in situ (FISH) de interfase a veces permite detectar una hiperdiploidía oculta.
La hiperdiploidía alta por lo general se presenta en los casos con factores clínicos de pronóstico favorable (pacientes de 1 a <10 años con recuento de glóbulos blancos [GB] bajo) y es, por sí sola, un factor independiente de pronóstico favorable. En un estudio, dentro del intervalo hiperdiploide de 51 a 65 cromosomas, los pacientes con números modales más altos (58–66), presentaron el mejor pronóstico. Las células leucémicas hiperdiploides son especialmente susceptibles a la apoptosis y acumulan concentraciones más altas de metotrexato y sus metabolitos activos de poliglutamato, lo que quizás explique el desenlace favorable que se observa con frecuencia en estos casos.
Aunque el desenlace general de los pacientes con hiperdiploidía alta se considera favorable, se ha observado que factores como la edad, el recuento de GB, las trisomías específicas y la respuesta temprana al tratamiento modifican su importancia pronóstica.
La importancia pronóstica de las trisomías cromosómicas específicas entre los niños con LLA-B hiperdiploide se ha descrito en múltiples informes.
- En un estudio en el que se combinó la experiencia del Children's Cancer Group y del Pediatric Oncology Group (POG) se observó que los pacientes con trisomías de los cromosomas 4, 10 y 17 (trisomías triples) tienen un desenlace particularmente favorable.; [Nivel de evidencia B4]
- En un informe en el que se usaron los datos del POG se encontró que los pacientes de riesgo estándar del NCI que exhiben trisomías 4 y 10 tienen un pronóstico excelente, independientemente del estado del cromosoma 17. En la actualidad, los protocolos del COG usan trisomías dobles de los cromosomas 4 y 10 para definir la hiperdiploidía favorable.
- En un análisis retrospectivo se evaluó a los pacientes tratados en 2 ensayos UKALL consecutivos con el fin de identificar y validar un perfil para predecir el desenlace de la LLA-B con hiperdiploidía alta. Los investigadores definieron un grupo de riesgo bajo (alrededor del 80 % de los pacientes con hiperdiploidía alta) que se relacionó con un pronóstico más favorable. Los pacientes de riesgo bajo tenían trisomías de los cromosomas 17 y 18 o trisomía de uno de estos cromosomas con ausencia de trisomías de los cromosomas 5 y 20. Los demás pacientes se definieron como de riesgo alto y tuvieron un desenlace menos favorable. La ERM al final de la inducción y las alteraciones del número de copias (como la deleción de IKZF1) fueron significativas para el pronóstico dentro de cada grupo de riesgo hiperdiploide.
Es posible que se encuentren translocaciones cromosómicas en combinación con hiperdiploidía alta; en estos casos, la clasificación de riesgo más apropiada para los pacientes se basa en la importancia pronóstica de la translocación. Por ejemplo, en un estudio, el 8 % de los pacientes con la fusión BCR::ABL1 también presentaban hiperdiploidía alta, y el desenlace de estos pacientes (tratados sin inhibidores de tirosina–cinasas) fue inferior al que se observó en los pacientes con hiperdiploidía alta negativos para BCR::ABL1.
Algunos pacientes con LLA hiperdiploide tienen un clon hipodiploide que se ha duplicado (hipodiploidía oculta). Las tecnologías moleculares, como las micromatrices de polimorfismos de un solo nucleótido que se usan para detectar la pérdida de heterocigosidad generalizada, se usan para identificar pacientes con hipodiploidía oculta. Estos casos se pueden interpretar de acuerdo con el perfil de ganancias y pérdidas de cromosomas específicos (hiperdiploidía con 2 o 4 copias de cromosomas en lugar de 3 copias). Estos pacientes tienen un desenlace desfavorable, similar al de aquellos con hipodiploidía.
La casi triploidía (68–80 cromosomas) y la casi tetraploidía (>80 cromosomas) son mucho menos comunes y son biológicamente diferentes de la hiperdiploidía alta. A diferencia de la hiperdiploidía alta, una gran proporción de casos con casi tetraploidía albergan una fusión ETV6::RUNX1 críptica. Antes se consideraba que la casi triploidía y tetraploidía estaban relacionadas con un pronóstico desfavorable, pero en estudios posteriores se indicó que es posible que no sea el caso.
El panorama genómico de la LLA hiperdiploide se caracteriza por variantes de los genes de la vía de receptores de tirosina–cinasas (RTK)/RAS en alrededor de la mitad de los casos. Los genes que codifican modificadores de histonas también se presentan de manera recurrente en una minoría de los casos. En el análisis de los perfiles de variantes se observa que las ganancias cromosómicas son episodios iniciales en la patogénesis de la LLA hiperdiploide y quizás se presente in utero, mientras que las variantes de los genes de la vía RTK/RAS son eventos tardíos en la leucemogénesis y por lo general son subclonales.
- Hipodiploidía (<44 cromosomas).
Los casos de LLA-B con un número de cromosomas menor que lo normal se subdividen de varias formas; en un informe se estratifican a partir del número modal de cromosomas en los siguientes cuatro grupos:
- Casi haploide: 24 a 29 cromosomas (n = 46).
- Hipodiploidía baja: 33 a 39 cromosomas (n = 26).
- Hipodiploidía alta: 40 a 43 cromosomas (n = 13).
- Casi diploide: 44 cromosomas (n = 54).
En los pacientes pediátricos con LLA-B, los casos casi haploides representan alrededor del 2 % de los casos de riesgo estándar del NCI y del 2 % de los casos de riesgo alto del NCI.
En los pacientes pediátricos con LLA-B, los casos de hipodiploidía baja representan alrededor del 0,5 % de los casos de riesgo estándar del NCI y del 2,6 % de los casos de riesgo alto del NCI.
La mayoría de pacientes con hipodiploidía se ubican en el grupo casi haploide o en el grupo de hipodiploidía baja; ambos grupos tienen un riesgo elevado de fracaso del tratamiento en comparación con los casos sin hipodiploidía. Los pacientes con menos de 44 cromosomas en sus células leucémicas tienen un desenlace más precario que aquellos con 44 a 45 cromosomas. En varios estudios se observó que los pacientes con enfermedad residual mínima (ERM) alta (≥0,01 %) después de la inducción evolucionan mal, con tasas de supervivencia sin complicaciones (SSC) a 5 años que oscilan entre el 25 % y el 47 %. Aunque la evolución es mejor para los pacientes con hipodiploidía que presentan una ERM baja después de la inducción (tasas de SSC a 5 años, 64–75 %), sus desenlaces siguen siendo inferiores a los de la mayoría de niños con otros tipos de LLA.
Las alteraciones genómicas recurrentes de la LLA casi haploide y con hipodiploidía baja son diferentes entre sí y de las de otros tipos de LLA. En la LLA casi haploide, son comunes las alteraciones que afectan la señalización RTK, la señalización RAS y el gen IKZF3. En la LLA con hipodiploidía baja, son comunes las alteraciones genéticas que afectan los genes TP53, RB1 y IKZF2. Es importante destacar que las alteraciones de TP53, que se observan en la LLA con hipodiploidía baja, también están presentes en las células no tumorales en alrededor del 40 % de los casos; esto indica que estas variantes son de la línea germinal y que la LLA con hipodiploidía baja representa, en algunos casos, una manifestación del síndrome de Li-Fraumeni. Cerca de dos tercios de los pacientes de LLA con variantes patógenas de la línea germinal en TP53 tienen LLA hipodiploide.
- Hiperdiploidía alta (51–65 cromosomas).
- Translocaciones cromosómicas y ganancias o deleciones de segmentos cromosómicos.
- Fusión ETV6::RUNX1 (t(12;21)(p13.2;q22.1)).
En los pacientes pediátricos con LLA-B, la fusión del gen ETV6 en el cromosoma 12 con el gen RUNX1 en el cromosoma 21 se presenta en alrededor del 27 % de los casos de riesgo estándar del NCI y del 10 % de los casos de riesgo alto del NCI.
La fusión ETV6::RUNX1 produce una translocación críptica que se detecta por métodos como la FISH, pero no por las pruebas citogenéticas convencionales; y se presenta de manera más frecuente en niños de 2 a 9 años. Los niños hispanos con LLA tienen una incidencia más baja de fusiones ETV6::RUNX1 que los niños blancos.
Por lo general, en los informes se indican tasas de SSC y supervivencia general (SG) favorables para los niños con la fusión ETV6::RUNX1; sin embargo, los siguientes factores modifican la repercusión pronóstica de esta característica genética:; [Nivel de evidencia B4]
- Respuesta temprana al tratamiento.
- Categoría de riesgo según el NCI (edad y recuento de GB en el momento del diagnóstico).
- Régimen de tratamiento.
En un estudio sobre el tratamiento de niños con diagnóstico nuevo de LLA, el análisis multivariante de los factores pronósticos indicó que la edad y el recuento leucocitario fueron factores pronósticos independientes, pero no el estado de la fusión ETV6::RUNX1. Sin embargo, en otro ensayo numeroso solo se inscribieron pacientes con LLA-B clasificada como de riesgo bajo, con características clínicas de riesgo bajo como trisomías de 4, 10 y 17 o fusión ETV6::RUNX1, y menos del 0,01 % de ERM al final de la inducción. Los pacientes tuvieron una tasa de remisión completa continua a 5 años del 93,7 % y una tasa de SG a 6 años del 98,2 % para los pacientes con ETV6::RUNX1. La presencia de anomalías citogenéticas secundarias, como la deleción de ETV6 (12p) o CDKN2A/B (9p), no parece afectar el desenlace de los pacientes con la fusión ETV6::RUNX1.
Las recaídas tardías son más frecuentes en los pacientes con fusiones ETV6::RUNX1 en comparación con otros casos de LLA-B recidivante. Los pacientes que exhiben la fusión ETV6::RUNX1 y recaen tienen un pronóstico un poco mejor que otros pacientes en recaída, el pronóstico es en particular favorable para los pacientes que recaen después de 36 meses del diagnóstico. Algunas recaídas en pacientes con fusiones ETV6::RUNX1 quizás indiquen la presencia de una lesión secundaria independiente en un clon preleucémico persistente (la lesión inicial sería la translocación ETV6::RUNX1).
- Fusión BCR::ABL1 (t(9;22)(q34.1;q11.2); Ph+).
La fusión BCR::ABL1 conduce a la producción de una proteína de fusión BCR::ABL1 con actividad de tirosina–cinasas (consultar la Figura 4). En los pacientes pediátricos con LLA-B, la fusión BCR::ABL1 se produce en alrededor del 2 % de los casos de riesgo estándar del NCI y del 5 % de los casos de riesgo alto del NCI. La fusión BCR::ABL1 también es un iniciador leucemógeno de la leucemia mieloide crónica (LMC). El punto de ruptura de BCR más común en la LMC es diferente del punto de ruptura de BCR más común en la LLA. Los puntos de ruptura típicos de la LMC producen una proteína de fusión más grande (llamada p210) que la proteína que producen los puntos de ruptura que se observan con mayor frecuencia en la LLA (llamada p190, una proteína de fusión más pequeña).
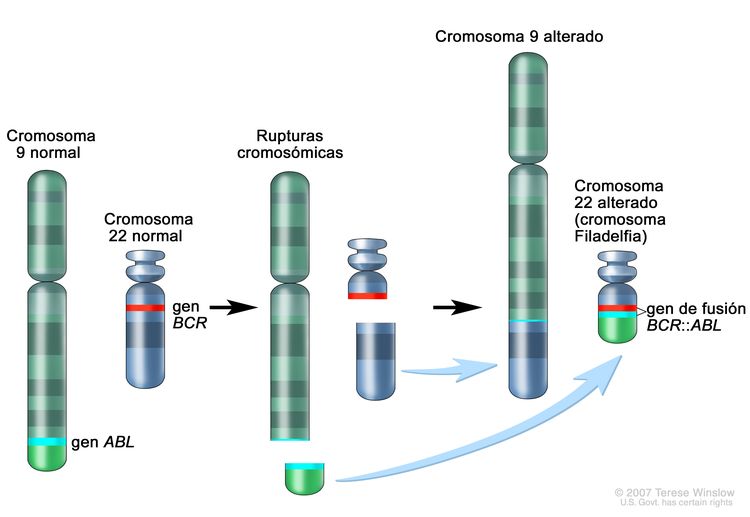 Figura 4. El cromosoma Filadelfia es una translocación entre el oncogén ABL1 (en el brazo largo del cromosoma 9) y el gen BCR (en el brazo largo del cromosoma 22), que produce el gen de fusión BCR::ABL1. La fusión BCR::ABL1 codifica una proteína oncogénica con actividad de tirosina–cinasas.
Figura 4. El cromosoma Filadelfia es una translocación entre el oncogén ABL1 (en el brazo largo del cromosoma 9) y el gen BCR (en el brazo largo del cromosoma 22), que produce el gen de fusión BCR::ABL1. La fusión BCR::ABL1 codifica una proteína oncogénica con actividad de tirosina–cinasas.La LLA Ph+ es más común en niños de más edad con LLA-B y recuentos de GB altos; la incidencia de las fusiones BCR::ABL1 aumenta a cerca del 25 % en adultos jóvenes con LLA.
Tradicionalmente, la fusión BCR::ABL1 se relacionó con un pronóstico muy adverso (en especial, en aquellos con un recuento de GB alto en el momento del diagnóstico, o con una respuesta temprana lenta al tratamiento inicial) y su presencia se ha considerado una indicación para el trasplante de células madres hematopoyéticas (TCMH) alogénico durante la primera remisión. Los inhibidores de tirosina–cinasas de BCR::ABL1, como el mesilato de imatinib, son eficaces en los pacientes con LLA con fusión BCR::ABL1. En un estudio del Children's Oncology Group (COG), en el que se administró quimioterapia intensiva y mesilato de imatinib simultáneo cada día, se observó una tasa de SSC a 5 años del 70 % (± 12 %), que fue superior a la tasa de SSC de los controles históricos de la era anterior a los inhibidores de tirosina–cinasas (mesilato de imatinib). Este resultado eliminó la recomendación de TCMH para pacientes con una buena respuesta temprana a la quimioterapia con inhibidores de tirosina–cinasas.
La International Consensus Classification de la leucemia o linfoma linfoblásticos de 2022 divide la LLA-B con fusión BCR::ABL1 en dos subtipos: casos con compromiso linfoide solo y casos con compromiso de varios linajes. Estos subtipos difieren en el momento del evento de transformación. Una célula progenitora multipotente es la célula de origen de la LLA-B con fusión BCR::ABL1 y compromiso de varios linajes, y una célula progenitora en estadio posterior es la célula de origen de la LLA-B con fusión BCR::ABL1 que solo compromete el linaje linfoide.
- La LLA-B con fusión BCR::ABL1 y compromiso linfoide solo es el subtipo predominante. Solamente una minoría de los casos en niños y adultos exhibe compromiso de varios linajes (estimado, 15 %–30 %).
- Los casos de LLA-B con fusión BCR::ABL1 y compromiso linfoide solo son similares a los casos con compromiso de varios linajes en aspectos como cuadro clínico inicial e inmunofenotipo. Además, ambos subtipos suelen exhibir la proteína de fusión p190.
- Una manera de diferenciar los pacientes con compromiso linfoide solo de los pacientes con compromiso de varios linajes es mediante la detección de la fusión BCR::ABL1 en células B, T y mieloides no afectadas por LLA.
- Otra forma de diferenciar entre los pacientes con compromiso linfoide solo o compromiso de varios linajes es mediante la detección de diferencias cuantitativas en las concentraciones de ERM (por lo general, 1 log) con métodos que cuantifican DNA o RNA con la fusión BCR::ABL1 y métodos que se basan en citometría de flujo, reacción en cadena de la polimerasa (PCR) cuantitativa en tiempo real o secuenciación de última generación (NGS) para la medición de los reordenamientos de inmunoglobulina (IG) o del receptor de células T (TCR) específicos de la leucemia.
- En pacientes de LLA-B con fusión BCR::ABL1 y compromiso linfoide solo, los cálculos de ERM mediante estos métodos se correlacionan entre sí.
- En pacientes de LLA-B con fusión BCR::ABL1 y compromiso de varios linajes, los cálculos de ERM después del tratamiento que se basan en la detección de DNA o RNA con la fusión BCR::ABL1 a menudo darán resultados más altos que la citometría de flujo o la cuantificación de los reordenamientos de IG o TCR específicos de la leucemia.
- En los pacientes de LLA-B con fusión BCR::ABL1 y compromiso de varios linajes, las concentraciones de trascriptos de BCR::ABL1 y DNA a veces permanecen estables en el tiempo a pesar de continuar el tratamiento con quimioterapia e inhibidores de tirosina–cinasas. En estas circunstancias, la persistencia de DNA o RNA con la fusión BCR::ABL1 posiblemente indique la presencia de un clon preleucémico, no de células leucémicas. Por lo tanto, el término ERM es inapropiado.
- Una conclusión sobre la diferencia en la detección de la ERM mediante métodos basados en medición del DNA o RNA que exhibe la fusión BCR::ABL1 versus la detección de la ERM por métodos de citometría de flujo o reordenamientos de IG o TCR es que ésta última opción es el método que produce el pronóstico más confiable. Por ejemplo, la presencia de ERM medida por detección de DNA o RNA con la fusión BCR::ABL1 en ausencia de detección de ERM por reordenamientos de IG o TCR no confiere un pronóstico más precario.
- A partir del número escaso de pacientes que se han estudiado hasta el momento, el pronóstico es semejante en adultos y niños con LLA-B con fusión de BCR::ABL1 y compromiso linfoide solo o compromiso de varios linajes.
- Hay informes de casos de pacientes con LLA-B con fusión BCR::ABL1 y compromiso de varios linajes que recaen años después del diagnóstico inicial. Además, su recaída presenta el mismo punto de ruptura de la fusión BCR::ABL1 que la leucemia inicial, pero presentan un reordenamiento diferente de IG o TCR. Estos informes de casos indican que los pacientes de LLA-B con fusión BCR::ABL1 y compromiso de varios linajes están en riesgo de un segundo evento leucemógeno que conlleva a otra leucemia con fusión BCR::ABL1.
- No hay evidencia de que un plan de vigilancia específico o el tratamiento prolongado con un inhibidor de tirosina–cinasas produzca un beneficio clínico en pacientes de LLA-B con fusión BCR::ABL1 y compromiso de varios linajes que mantienen la expresión de trascriptos BCR::ABL1 o DNA con esta fusión en el momento de terminar el tratamiento estándar de la leucemia.
- LLA con reordenamiento de KMT2A (t(v;11q23.3)).
Los reordenamientos que afectan el gen KMT2A con más de 100 genes compañeros de translocación producen oncoproteínas de fusión. Los reordenamientos en el gen KMT2A se presentan hasta en el 80 % de los lactantes con LLA. En los pacientes pediátricos de más de un año de edad con LLA-B, cerca del 1 % de los casos de alto riesgo estándar del NCI y del 4 % de los casos de riesgo alto del NCI tienen reordenamientos de KMT2A.
Estos reordenamientos se suelen relacionar con aumento del riesgo de fracaso del tratamiento, sobre todo en lactantes. En los niños con LLA, la fusión KMT2A::AFF1 (t(4;11)(q21;q23)) es el reordenamiento más común que afecta el gen KMT2A y se presenta en el 1 % al 2 % de los casos.
Los pacientes con fusiones KMT2A::AFF1 por lo general son lactantes con recuentos de GB altos. Estos pacientes son más propensos que otros niños con LLA a presentar enfermedad en el sistema nervioso central (SNC) y una respuesta precaria al tratamiento inicial. Si bien, tanto los lactantes como los adultos con la fusión KMT2A::AFF1 tienen un riesgo alto de fracaso del tratamiento, los niños con esta fusión tienen mejores desenlaces. Con independencia del tipo de reordenamiento del gen KMT2A, los lactantes con LLA que exhibe reordenamientos del gen KMT2A presentan tasas de supervivencia sin complicaciones mucho más desfavorables que los pacientes pediátricos de más de 1 año con LLA que exhibe reordenamientos de KMT2A.
Mediante secuenciación de genoma completo se determinó que los casos de LLA infantil con reordenamientos del gen KMT2A tienen variantes subclonales frecuentes de NRAS o KRAS y pocas anomalías genómicas adicionales, ninguna de importancia clínica bien definida. La deleción del gen KMT2A no se ha relacionado con un pronóstico adverso.
Como dato interesante, la fusión KMT2A::MLLT1 (t(11;19)(q23;p13.3)) se presenta en alrededor del 1 % de los casos de LLA, tanto en la LLA de linaje B temprano como en la LLA-T. El desenlace de los lactantes con la fusión KMT2A::MLLT1 es precario, pero es relativamente favorable en los niños de más edad con LLA-T que presentan esta fusión.
- Fusión TCF3::PBX1 (t(1;19)(q23;p13.3)) y fusión TCF3::HLF (t(17;19)(q22;p13)).
En los pacientes pediátricos con LLA-B, la fusión del gen TCF3 en el cromosoma 19 con el gen PBX1 en el cromosoma 1 se presenta en alrededor del 4 % de los casos de riesgo estándar del NCI y del 5 % de los casos de riesgo alto del NCI. La fusión TCF3::PBX1 se presenta como una translocación equilibrada o desequilibrada, y es la principal anomalía genómica recurrente del inmunofenotipo de LLA pre-B (positiva para inmunoglobulina citoplasmática). Los niños negros son más propensos a presentar LLA pre-B con la fusión TCF3::PBX1 que los niños blancos.
La fusión TCF3::PBX1 se relacionó con un desenlace inferior en el contexto de un tratamiento a base de antimetabolitos, pero la importancia para determinar un pronóstico adverso se invalidó casi por completo cuando se usó un tratamiento multifarmacológico más intensivo. En particular, en un ensayo realizado por el St. Jude Children's Research Hospital (SJCRH) en el que todos los pacientes se trataron sin radiación craneal, aquellos con la fusión TCF3::PBX1 tuvieron un desenlace general comparable al de los niños sin esta translocación, pero presentaron un riesgo más alto de recaída en el SNC y una tasa más baja de recaída en la médula ósea; esto indica que quizás estos pacientes necesiten un tratamiento dirigido al SNC más intensivo.
La fusión TCF3::HLF se presenta en menos del 1 % de los casos de LLA infantil. La LLA con la fusión TCF3::HLF se relaciona con coagulación intravascular diseminada e hipercalcemia en el momento del diagnóstico. El desenlace es muy precario en niños con la fusión TCF3::HLF: en una revisión de la literatura se indicó una mortalidad de 20 de 21 casos notificados. Además de la fusión TCF3::HLF, el panorama genómico de este subtipo de LLA se caracterizó por deleciones en genes que participan en el desarrollo de las células B (PAX5, BTG1 y VPREB1) y por variantes de los genes de la vía RAS (NRAS, KRAS y PTPN11).
- LLA con reordenamiento de DUX4 y deleciones de ERG frecuentes.
En los pacientes pediátricos con LLA-B, casi el 3 % de los casos de riesgo estándar del NCI y del 6 % de los casos de riesgo alto del NCI tienen un reordenamiento que afecta el gen DUX4 y conduce a su sobreexpresión. Tener ascendencia de Asia oriental se relacionó con un aumento de la prevalencia de LLA con reordenamiento de DUX4 (favorable). El reordenamiento más común produce fusiones IGH::DUX4; y también se observan fusiones ERG::DUX4. Los casos con reordenamiento de DUX4 exhiben un perfil de expresión génica característico que al principio se identificó como asociado con deleciones focales en ERG, y entre la mitad y más de dos tercios de estos casos tienen deleciones intragénicas focales que afectan el gen ERG pero que no se encuentran en otros subtipos de LLA. Las deleciones de ERG a menudo parecen ser clonales, pero al usar métodos de detección más sensibles, se observa que la mayoría de los casos son policlonales. Se observan alteraciones de IKZF1 en el 20 % al 40 % de los casos de LLA con reordenamiento de DUX4.
La deleción de ERG conlleva un pronóstico excelente, con tasas de SG superiores al 90 %. El pronóstico sigue siendo favorable incluso cuando hay una deleción de IZKF1. Si bien los pacientes de LLA con reordenamiento de DUX4 tienen un pronóstico general favorable, no se sabe si esto es cierto en los casos con deleción de ERG y en los casos con ERG intacto. En un estudio de 50 pacientes de LLA con reordenamiento de DUX4, los pacientes con deleción de ERG detectada mediante PCR (n = 33) presentaron una tasa de SSC más favorable, de alrededor del 90 %, que los pacientes con ERG intacto (n = 17), con una tasa de SSC de alrededor del 70 %.
- LLA con reordenamiento de MEF2D.
En los pacientes pediátricos con LLA-B, las fusiones génicas que afectan a MEF2D, un factor de transcripción que se expresa durante el desarrollo de las células B, se observan en alrededor del 0,3 % de los casos de riesgo estándar del NCI y del 3 % de los casos de riesgo alto del NCI.
Aunque hay múltiples compañeros de fusión, la mayoría de los casos afectan el gen BCL9, en el cromosoma 1q21, al igual que el gen MEF2D. La deleción intersticial que produce la fusión MEF2D::BCL9 es muy pequeña como para ser detectada por métodos citogenéticos convencionales. Los casos con fusiones del gen MEF2D exhiben un perfil de expresión génica característico, excepto por casos infrecuentes que tienen MEF2D::CSFR1 y un perfil de expresión génica similar a BCR::ABL1.
La mediana de edad en el momento del diagnóstico para los casos de LLA con reordenamiento de MEF2D fue de 12 a 14 años en los estudios que incluyeron pacientes adultos y niños. En los 22 niños de LLA con reordenamiento de MEF2D inscritos en un ensayo clínico de LLA de riesgo alto, la tasa de SSC a 5 años fue del 72 % (error estándar, ±10 %), que fue inferior a la de otros pacientes.
- LLA con reordenamiento de ZNF384.
En los pacientes pediátricos con LLA-B, los reordenamientos en el gen ZNF384, que codifica un factor de transcripción, se presentan en cerca del 0,3 % de los casos de riesgo estándar del NCI y del 2,7 % de los casos del riesgo alto del NCI.
Tener ascendencia de Asia oriental se relacionó con un aumento en la prevalencia de ZNF384. Se han descrito múltiples compañeros de fusión para ZNF384, entre ellos, ARID1B, CREBBP, EP300, SMARCA2, TAF15 y TCF3. Sin importar el compañero de fusión, los casos de LLA con reordenamiento de ZNF384 exhiben un perfil de expresión génica característico. El reordenamiento de ZNF384 no parece tener importancia pronóstica independiente. Sin embargo, dentro del subgrupo de pacientes con reordenamientos de ZNF384, los pacientes con fusiones EP300::ZNF384 tienen tasas de recaídas más bajas que los pacientes con fusiones de ZNF384 en las que intervienen otros compañeros de fusión. El inmunofenotipo de LLA-B con reordenamiento de ZNF384 se caracteriza por expresión débil de CD10 o ausencia de expresión de este producto, mientras que es común que exprese CD13 o CD33. Se han notificado casos de leucemia aguda de fenotipo mixto (LAFM) (B/mieloide) que exhiben fusiones génicas de ZNF384, y en la evaluación genómica de la LAFM se encontró que las fusiones génicas de ZNF384 estaban presentes en cerca de la mitad de los casos con fenotipo B/mieloide.
- LLA-B con reordenamiento de NUTM1.
La LLA-B con reordenamiento de NUTM1 se observa con más frecuencia en lactantes, y representa el 3 % al 5 % de todos los casos de LLA-B en ese grupo etario y alrededor del 20 % de los casos de lactantes con LLA-B sin reordenamiento de KMT2A. La frecuencia del reordenamiento de NUTM1 es más baja en los niños después del primer año de vida (<1% % de casos).
El gen NUTM1 se encuentra en el cromosoma 15q14, y algunos casos de LLA-B con reordenamientos de NUTM1 presentan anomalías en el cromosoma 15q, pero otros son casos crípticos y no presentan anomalías citogenéticas. La secuenciación del RNA, así como la técnica FISH con sondas de separación, se usan para detectar la presencia de reordenamiento de NUTM1.
El reordenamiento de NUTM1 está relacionado con un desenlace favorable. De 35 lactantes con LLA-B con reordenamiento de NUTM1 que se trataron en los protocolos Interfant, todos los pacientes lograron la remisión y no se observaron recaídas. En los 32 niños mayores de 12 meses que presentaban LLA-B con reordenamiento de NUTM1, las tasas de SSC a 4 años y de SG fueron del 92 % y del 100 %, respectivamente.
- Fusión IGH::IL3 (t(5;14)(q31.1;q32.3)).
Esta entidad se incluyó en la revisión de 2016 de la clasificación de tumores de tejidos hematopoyéticos y linfoides de la Organización Mundial de la Salud (OMS). El hallazgo de la t(5;14)(q31.1;q32.3) en pacientes con LLA e hipereosinofilia en la década de 1980 fue seguido por la identificación de la fusión IGH::IL3 como la causa genética subyacente de esta afección. La unión del locus de IGH a la región promotora del gen IL3 lleva a la desregulación de la expresión de IL3. Las anomalías citogenéticas en niños con LLA y eosinofilia son variables, solo un subgrupo se produce a partir de la fusión IGH::IL3.
El número de casos de LLA con IGH::IL3 descritos en la bibliografía publicada es demasiado pequeño como para evaluar la importancia pronóstica de la fusión IGH::IL3. El diagnóstico de los casos de LLA con IGH::IL3 quizá se retrase porque el clon de LLA en la médula ósea a veces es pequeño y porque es posible que se presente con hipereosinofilia en ausencia de citopenias y blastocitos circulantes.
- Amplificación intracromosómica del cromosoma 21 (iAMP21).
La iAMP21 se presenta en alrededor del 5 % de los casos pediátricos de LLA-B del NCI de riesgo estándar y el 7 % de los casos del NCI de riesgo alto. Por lo general la iAMP21 se diagnostica mediante FISH y se define por la presencia de 5 o más señales de RUNX1 por célula (o ≥3 copias adicionales de RUNX1 en un cromosoma anormal único). La iAMP21 también se puede identificar mediante análisis de micromatriz cromosómica. Rara vez, la iAMP21 con un patrón genómico atípico (por ejemplo, amplificación de la región genómica, pero con menos de 5 señales RUNX1 o que tiene al menos 5 señales RUNX1 con algunas separadas del cromosoma iAMP21 anormal) se identifica mediante micromatriz, pero no con FISH de RUNX1. No se ha descrito la importancia pronóstica de iAMP21 definida solo mediante micromatriz.
La iAMP21 se relaciona con mayor edad (mediana, alrededor 10 años), recuento de GB inferior a 50 × 109/l, un leve predominio en mujeres y una ERM alta al final de la inducción. El análisis de las firmas de variantes indican que las amplificaciones génicas en iAMP21 se presentan más tarde en la leucemogénesis, lo cual contrasta con las de la LLA hiperdiploide que pueden presentarse temprano en la vida e incluso in utero.
El grupo de ensayos clínicos United Kingdom Acute Lymphoblastic Leukaemia (UKALL), inicialmente notificó que la presencia de iAMP21 confirió un pronóstico precario a los pacientes tratados en el ensayo MRC ALL 97/99 (tasa de SSC a 5 años, 29 %). En el ensayo posterior del mismo grupo (UKALL2003 [NCT00222612]), se asignó a los pacientes con iAMP21 a recibir un régimen de quimioterapia más intensivo, y estos pacientes presentaron un desenlace mucho más favorable (tasa de SSC a 5 años, 78 %). De manera similar, el COG informó que la iAMP21 se relacionó con un desenlace significativamente inferior en los pacientes de riesgo estándar del NCI (tasa de SSC a 4 años, 73 % para iAMP21 vs. 92 % en otros), pero no en los pacientes con riesgo alto del NCI (tasa de SSC a 4 años, 73 % vs. 80 %). En un análisis multivariante, la iAMP21 fue un factor de pronóstico independiente de desenlace precario solo en los pacientes de riesgo estándar del NCI. Los resultados de los estudios UKALL2003 y COG indican que en los pacientes que tienen una iAMP21, el tratamiento con regímenes de quimioterapia de riesgo alto anula la repercusión de iAMP21 como factor de pronóstico adverso y evita la necesidad de un TCMH en el momento de la primera remisión.
- Alteraciones en PAX5.
En análisis de expresión génica se identificaron dos subtipos de LLA diferenciados con alteraciones genómicas en PAX5, denominadas PAX5alt y PAX5 p.P80R (NP_057953.1). Las alteraciones en el subtipo PAX5alt incluyen reordenamientos, variantes de secuencia y amplificaciones intragénicas focales.
PAX5alt. En los pacientes pediátricos con LLA-B, se han notificado reordenamientos de PAX5 en alrededor del 3 % de los casos de riesgo estándar del NCI y del 11 % de los casos de riesgo alto del NCI. Se identificaron más de 20 genes compañeros de PAX5,. PAX5::ETV6, la alteración genómica primaria en dic(9;12)(p13;p13), fue la fusión génica más común.
Se identificó una amplificación intragénica de PAX5 en cerca del 1 % de los casos de LLA-B, y por lo general se detectó en casos que no tenían alteraciones genómicas que se sabe son iniciadoras de leucemia. Los casos con amplificación de PAX5 son de predominio masculino (66 %), y la mayoría (55 %) se clasifican en un estado de riesgo alto del NCI. En una cohorte de pacientes con amplificación de PAX5 que recibieron el diagnóstico entre 1993 y 2015, la tasa de SSC a 5 años fue del 49 % (intervalo de confianza [IC] 95 %, 36–61 %), y la tasa de SG fue del 67 % (IC 95 %, 54–77 %), lo que indica un pronóstico relativamente más precario para pacientes con este subtipo de LLA-B.
PAX5 p.P80R (NP_057953.1). La leucemia que tiene PAX5 con la variante p.P80R exhibe un perfil de expresión génica diferente al de otros casos con alteraciones en PAX5. En los pacientes pediátricos con LLA-B, los casos con PAX5 p.P80R representan alrededor del 0,3 % de los casos de riesgo estándar del NCI y del 1,8 % de los casos de riesgo alto del NCI. La LLA-B con PAX5 p.P80R se presenta con más frecuencia en los adolescentes y adultos jóvenes (AAJ) y en los adultos (3,1 % y 4,2 %, respectivamente).
Para los pacientes pediátricos con la variante p.P80R en PAX5 y con la variante PAX5alt tratados en los ensayos clínicos del COG, el desenlace es intermedio (SSC a 5 años, alrededor del 75 %). Los reordenamientos de PAX5alt también se detectaron en pacientes lactantes con LLA, y los desenlaces notificados fueron similares a los de los niños de LLA con reordenamiento de KMT2A.
- Similar a BCR::ABL1 (similar a Ph).
Los pacientes con un resultado negativo para BCR::ABL1 que exhiben un perfil de expresión génica semejante al de los pacientes con resultado positivo para BCR::ABL1 se considera que tienen una LLA similar a Ph, que ahora se conoce como similar a BCR::ABL1. Esto sucede en el 10 % al 20 % de los pacientes de LLA-B infantil; su frecuencia aumenta con la edad y se ha relacionado con una variante o deleción de IKZF1.
En análisis retrospectivos se indicó que los pacientes con LLA similar a BCR::ABL1 tienen un pronóstico precario. En una serie, la tasa de SSC a 5 años de los niños y adolescentes de riesgo alto según el NCI con LLA similar a BCR::ABL1 fue del 58 % y el 41 %, respectivamente. Si bien el subtipo similar a BCR::ABL1 es más frecuente en pacientes de más edad y de riesgo más alto, también se ha identificado en pacientes de riesgo estándar según el NCI. En un estudio del COG, se encontró que el 13,6 % de 1023 pacientes con LLA-B de riesgo estándar según el NCI tenían una LLA similar a BCR::ABL1; estos pacientes exhibieron una tasa de SSC inferior comparada con los pacientes de riesgo estándar con una LLA de otro tipo no similar a BCR::ABL1 (82 % vs. 91 %), aunque no se indicaron diferencias en la tasa de SG (93 % vs. 96 %). En un estudio de 40 pacientes con LLA similar a BCR::ABL1, la importancia pronóstica adversa de este subtipo se anuló cuando los pacientes recibieron tratamiento dirigido según el riesgo a partir de las concentraciones de ERM.
El sello distintivo de la LLA similar a BCR::ABL1 es la activación de la señalización de cinasa; alrededor del 35 % al 50 % exhibe alteraciones genómicas en CRLF2 y de esos, la mitad exhibe de manera simultánea variantes de JAK.
Se observó que en muchos de los casos restantes de LLA similar a BCR::ABL1 hay una serie de translocaciones que afectan a genes de fusión de clase ABL codificadores de tirosina–cinasas, como ABL1, ABL2, CSF1R, y PDGFRB. En algunos casos, se ha observado que las proteínas de fusión de estas combinaciones de genes son transformadoras y responden a los inhibidores de tirosina–cinasas in vitro e in vivo, lo que indica posibles estrategias terapéuticas para estos pacientes.
En los pacientes pediátricos con LLA-B, los casos de LLA similar a BCR::ABL1 con alteraciones genómicas que no afectan CRLF2 representan alrededor del 3 % de los casos de riesgo estándar del NCI y del 8 % de los casos de riesgo alto del NCI. En un estudio retrospectivo de 122 pacientes pediátricos (edad 1–18 años) con fusiones de clase ABL (todos recibieron tratamiento sin inhibidores de tirosina–cinasas), la tasa de SSC a 5 años fue del 59 % y la tasa de SG fue del 76 %.
Alrededor del 9 % de los casos de LLA similar a BCR::ABL1 se producen a partir de reordenamientos que llevan a la sobreexpresión de un receptor de eritropoyetina (EPOR) incompleto. La región del extremo C del receptor que se pierde es la región alterada en la policitemia congénita familiar primaria, y es la que controla la estabilidad de EPOR. La porción de EPOR que queda es suficiente para la activación de JAK-STAT y para conducir a la aparición de la leucemia. Las variantes puntuales de los genes de cinasas, distintos a JAK1 y JAK2, son poco comunes en los casos de LLA similar a BCR::ABL1.
CRLF2. Se han identificado alteraciones genómicas en CRLF2, un gen de un receptor de citocina ubicado en las regiones pseudoautosómicas de los cromosomas sexuales, en el 5 % al 10 % de los casos de LLA-B. Estas variaciones representan alrededor del 50 % de los casos de LLA similar a BCR::ABL1. Las anomalías cromosómicas que con mayor frecuencia conducen a la sobreexpresión de CRLF2 incluyen las translocaciones del locus de IGH (cromosoma 14) al CRLF2 y las deleciones intersticiales en las regiones pseudoautosómicas de los cromosomas sexuales, lo que produce una fusión P2RY8::CRLF2. Estas dos alteraciones genómicas se relacionan con características clínicas y biológicas diferenciadoras.
En los pacientes pediátricos con LLA-B, la LLA-B similar a BCR::ABL1 con alteraciones genómicas que afectan CRLF2 se observa en alrededor del 2 % de los casos de riesgo estándar del NCI y del 5 % de los casos de riesgo alto del NCI.
La LLA con variaciones genómicas en CRLF2 presenta una mayor incidencia en niños que tienen ascendencia genética hispana o latina, e indígena americana. En un estudio de 205 niños con LLA-B de riesgo alto, 18 de 51 (35,3 %) pacientes hispanos o latinos presentaron reordenamientos de CRLF2, en comparación con 11 casos de 154 (7,1 %) pacientes en el resto de las etnias manifestadas. En un segundo estudio, se observó que solo la frecuencia de fusiones IGH::CRLF2 estaba aumentada en los niños hispanos o latinos en comparación con los niños con LLA-B que no eran hispanos ni latinos (12 vs. 2,7 %). En este estudio, el porcentaje de LLA-B con fusiones P2RY8::CRLF2 fue de alrededor del 6 % y no se vio modificado por la etnia.
La fusión P2RY8::CRLF2 se observa en el 70 % al 75 % de los pacientes pediátricos con alteraciones genómicas en CRLF2, y se presenta en pacientes jóvenes (mediana de edad, 4 vs. 14 años en los pacientes con IGH::CRLF2). A menudo la P2RY8::CRLF2 se presenta junto a otras anomalías cromosómicas establecidas (por ejemplo, hiperdiploidía, iAMP21, dic(9;20)), por el contrario IGH::CRLF2 en general es mutuamente excluyente de los subtipos citogenéticos conocidos. Se observan alteraciones genómicas en CRLF2 en cerca del 60 % de los pacientes con LLA y síndrome de Down, y la fusión P2RY8::CRLF2 es más frecuente que la fusión IGH::CRLF2 (cerca del 80–85 % vs. 15–20 %).
La presencia de IGH::CRLF2 y P2RY8::CRLF2 a menudo es una alteración temprana en la formación de una LLA-B y exhibe prevalencia clonal. Sin embargo, en algunos casos es una alteración tardía y exhibe prevalencia subclonal. En estos casos, la pérdida de la anormalidad genómica de CRLF2 en el momento de una recaída confirma la naturaleza subclonal de esta alteración.
Las anormalidades en CRLF2 se asocian directamente con deleciones de IKZF1. Estas deleciones son más frecuentes en casos con fusiones IGH::CRLF2 que en casos con fusiones P2RY8::CRLF2. Otras alteraciones genómicas recurrentes asociadas con las alteraciones en CRLF2 son las deleciones en los genes vinculados con la diferenciación de las células B (por ejemplo, PAX5, BTG1, EBF1, etc.) y el control del ciclo celular (CDKN2A), así como las alteraciones genómicas que activan la vía de señalización JAK-STAT (por ejemplo, variantes de IL7R y JAK).
Aunque los resultados de varios estudios retrospectivos indican que las anomalías en CRLF2 quizás denoten un pronóstico adverso en los análisis univariantes, la mayoría no indica que esta anomalía sea un factor de predicción independiente del desenlace. Por ejemplo, en un estudio europeo grande, la expresión elevada de CRLF2 no se relacionó con un desenlace desfavorable en los análisis multivariantes, mientras que las firmas de expresión de deleción de IKZF1 y similar a BCR::ABL1 se relacionaron con desenlaces desfavorables. También hay polémica sobre si el análisis de la importancia pronóstica de las anomalías en CRLF2 debe hacerse en relación con la sobreexpresión de CRLF2 o con la presencia de anomalías genómicas en CRLF2.
- Deleciones de IKZF1.
Las deleciones de IKZF1, incluso las deleciones del gen completo y de exones específicos, están presentes en alrededor del 15 % de los casos de LLA-B. Con menor frecuencia, el gen IKZF1 se inactiva por variantes puntuales deletéreas.
Los casos con deleciones de IKZF1 se suelen presentar en niños mayores, que tienen un recuento de GB más alto en el momento del diagnóstico y, por lo tanto, son más frecuentes en los pacientes de riesgo alto según el NCI en comparación con los de riesgo estándar según el NCI. Una proporción alta de casos positivos para BCR::ABL1 tienen una deleción de IKZF1, y las LLA que surgen en niños con síndrome de Down tienen tasas elevadas de deleciones de IKZF1. Las deleciones de IKZF1 también son comunes en los casos con alteraciones genómicas de CRLF2 y en la LLA similar a BCR::ABL1.
En varios informes se ha documentado la relevancia de la deleción de IKZF1 para definir un pronóstico adverso, y, en la mayoría de los estudios con análisis multivariantes, se notificó que esta deleción es un factor independiente de pronóstico precario.; [Nivel de evidencia B4] Sin embargo, la importancia pronóstica de IKZF1 quizás no sea equivalente en todos los subtipos biológicos de LLA, como se demuestra por la aparente falta de relevancia pronóstica en los pacientes con deleciones de ERG. De manera parecida, la importancia pronóstica de la deleción de IKZF1 también se redujo en una cohorte de pacientes del COG de LLA con reordenamiento de DUX4 y desregulación transcripcional de ERG, lo que a menudo se presenta por la deleción de ERG. El grupo de la Associazione Italiana di Ematologia e Oncologia Pediatrica y el Berlin-Frankfurt-Münster notificó que las deleciones de IKZF1 indicaron un pronóstico adverso solo en los pacientes con LLA-B que exhibían ERM alta al final de la inducción y en quienes se detectaron al mismo tiempo deleciones en CDKN2A, CDKN2B, PAX5 o PAR1 (en ausencia de la deleción de ERG). El pronóstico precario relacionado con las alteraciones en IKZF1 empeora con el hallazgo concomitante de la deleción 22q11.22. En un estudio de 1310 pacientes con LLA-B, cerca de la mitad de los pacientes con alteraciones en IKZF1 también tenían una deleción 22q11.22. La tasa de SSC fue del 43,3 % para aquellos con las 2 anomalías, en comparación con el 68,5 % de los pacientes con alteraciones en IKZF1 y 22q11.22 natural (P< 0,001).
Hay pocos resultados publicados sobre el cambio de tratamiento a partir del estado del gen IKZF1. El grupo Malasia-Singapur publicó los resultados de dos ensayos consecutivos. En el primer ensayo (MS2003), el estado de IKZF1 no se consideró en la estratificación del riesgo, mientras que en el ensayo posterior (MS2010), los pacientes con deleción de IKZF1 se excluyeron del grupo de riesgo estándar. Además, en el ensayo MS2010, más pacientes con deleción de IKZF1 recibieron terapia intensificada. Los pacientes de LLA con deleción de IKZF1 exhibieron mejores desenlaces en el ensayo MS2010 en comparación con los pacientes del ensayo MS2003, pero la interpretación de esta observación se ve limitada por otros cambios en la estratificación del riesgo y las diferencias de tratamiento entre los dos ensayos.[Nivel de evidencia B4]
En el estudio holandés ALL11, los pacientes con deleciones en IKZF1 prolongaron su terapia de mantenimiento durante 1 año con el objetivo de mejorar el desenlace. En el análisis de referencia se demostró una reducción de casi 3 veces en la tasa de recidiva y una mejora en la tasa de SSC a 2 años (del 74,4 % al 91,2 %), en comparación con los controles históricos.
- LLA con reordenamiento de MYC (8q24).
Los reordenamientos del gen MYC son un hallazgo infrecuente pero recurrente en pacientes pediátricos con LLA-B. Se han notificado pacientes con reordenamientos del gen MYC, así como de los genes IGH2, IGK y IGL en 14q32, 2p12 y 22q11.2, respectivamente. Los linfoblastos por lo general exhiben un inmunofenotipo de células B precursoras, con una morfología francesa-americana-británica (FAB) L2 o L3, sin expresión de inmunoglobulina de superficie ni cadenas ligeras κ o λ. Se han observado reordenamientos simultáneos del gen MYC junto con otros reordenamientos citogenéticos como IGH::BCL2 o KMT2A. Los pacientes mencionados en la bibliografía, recibieron diversas terapias para la LLA o se trataron siguiendo los protocolos de linfoma o leucemia de células B maduras; sin embargo, todavía no está claro cuál es el mejor tratamiento para estos pacientes.
- Fusión ETV6::RUNX1 (t(12;21)(p13.2;q22.1)).
Características citogenéticas y genómicas de la leucemia linfoblástica aguda de células T
La LLA-T se caracteriza por alteraciones genómicas que producen la activación de programas transcripcionales relacionados con el desarrollo de las células T y por una frecuencia alta de casos (casi el 60 %) con variantes de NOTCH1 o FBXW7 que producen la activación de la vía NOTCH1. Las anomalías citogenéticas comunes en la LLA-B (por ejemplo, hiperdiploidía, 51–65 cromosomas) son poco frecuentes en la LLA-T.
En la Figura 5 de más abajo, los casos de LLA-T se dividen en 10 subtipos moleculares según la expresión del RNA y la presencia de variantes de los genes. Estos casos provienen de pacientes inscritos en los ensayos clínicos SJCRH y COG. Cada subtipo se relaciona con la desregulación de genes específicos que participan en la formación de las células T. Dentro de un subtipo, es posible que la expresión del gen desregulado se vea impulsada por múltiples mecanismos. Por ejemplo, en el subtipo más abundante, TAL1, es posible que la sobrexpresión de TAL1 sea el resultado de la fusión STIL::TAL1 y de la variante de inserción no codificante cadena arriba en el locus de TAL1 que crea un sitio de unión a MYB. A modo de otro ejemplo, dentro del grupo de HOXA, es posible que la sobrexpresión de HOXA9 sea el resultado de múltiples fusiones génicas, como los reordenamientos de KMT2A o MLLT10 y las fusiones SET::NUP214. A diferencia de los subtipos moleculares de LLA-B, los subtipos moleculares de LLA-T no se usan para definir los tratamientos según su importancia pronóstica o sus implicaciones terapéuticas.
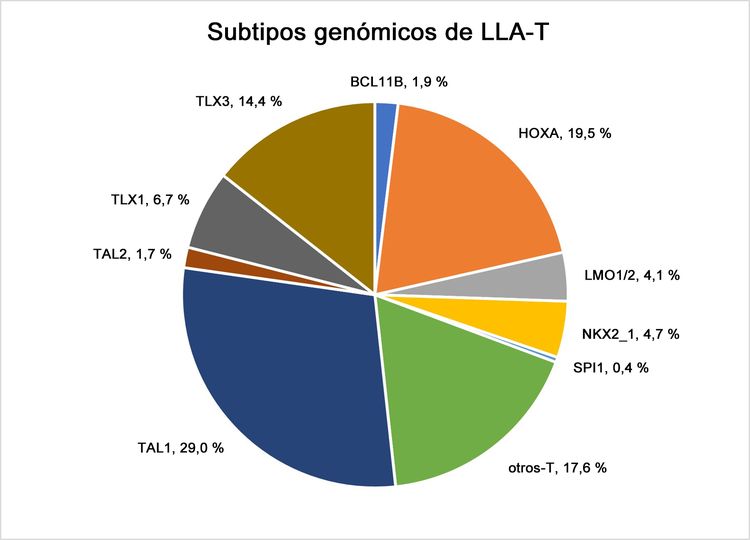 Figura 5. Subtipos genómicos de LLA-T. En la figura se observan los datos de 466 niños, adolescentes y adultos jóvenes diagnosticados con LLA-T e inscritos en los ensayos clínicos del St. Jude Children’s Research Hospital o el Children’s Oncology Group. Adaptado de Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
Figura 5. Subtipos genómicos de LLA-T. En la figura se observan los datos de 466 niños, adolescentes y adultos jóvenes diagnosticados con LLA-T e inscritos en los ensayos clínicos del St. Jude Children’s Research Hospital o el Children’s Oncology Group. Adaptado de Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
- Señalización de la vía Notch.
La señalización de la vía Notch a menudo se activa por variantes de los genes NOTCH1 y FBXW7 en casos de LLA-T, y estos son los genes alterados con mayor frecuencia en los casos de LLA-T infantil. Las variantes que activan el gen NOTCH1 se presentan en cerca del 50 % al 60 % de los casos de LLA-T; las variantes que inactivan el gen FBXW7 se presentan en cerca del 15 % de los casos. Casi el 60 % de los casos de LLA-T exhiben activación de la vía Notch por variantes de por lo menos uno de estos genes.
La importancia pronóstica de las variantes de NOTCH1 y FBXW7 quizás esté modulada por alteraciones genómicas en RAS y PTEN. El French Acute Lymphoblastic Leukaemia Study Group (FRALLE) y el Group for Research on Adult Acute Lymphoblastic Leukemia notificaron que los pacientes con variantes de NOTCH1 o FBXW7 que además tienen tipos naturales de PTEN y RAS forman un grupo de pronóstico favorable (es decir, riesgo bajo), mientras que los pacientes con variantes de PTEN o RAS, sin importar el estadio de NOTCH1 y FBXW7, tienen un riesgo significativamente más alto de fracaso del tratamiento (es decir, grupo de riesgo alto). En el estudio FRALLE, la tasa de supervivencia sin enfermedad a 5 años fue del 88 % para el grupo de pacientes de riesgo genético bajo y del 60 % para el grupo de pacientes de riesgo genético alto. Sin embargo, al usar los mismos criterios para definir el grupo de riesgo genético, no fue posible que el consorcio Dana-Farber Cancer Institute replicara estos resultados. Ellos informaron una tasa de SSC a 5 años del 86 % para los pacientes de riesgo genético bajo y del 79 % para los pacientes de riesgo genético alto, una diferencia que no fue estadísticamente significativa (P = 0,26).
- Translocaciones cromosómicas.
En la LLA-T se han identificado múltiples translocaciones cromosómicas que llevan a la alteración en la expresión de genes específicos. Estos reordenamientos cromosómicos producen fusiones de genes codificadores de factores de transcripción (por ejemplo, TAL1, TAL2, LMO1, LMO2, LYL1, TLX1, TLX3, NKX2-I, HOXA y MYB) con uno de los locus de los receptores de las células T (o con otros genes), lo que lleva a la alteración en la expresión de los factores de transcripción en las células leucémicas. A menudo, estas translocaciones no son evidentes al examinar el cariotipo estándar, pero se logran confirmar con técnicas de detección más sensibles, como FISH o PCR. Las variantes de una región no codificante cerca del gen TAL1 que produce un superpotenciador antes de la secuencia del gen TAL1 representan alteraciones genómicas que no son translocaciones, pero que también activan la transcripción de TAL1 e inducen la LLA-T.
En la LLA-T también se observan translocaciones que producen proteínas de fusión quiméricas.
- Se ha observado una fusión NUP214::ABL1 en el 4 % al 6 % de los casos de LLA-T en adultos y niños, con predomino masculino. La fusión es críptica en el análisis citogenético, y se observa en la FISH en los episomas amplificados, o con menor frecuencia, como una región pequeña de tinción homogénea. En casos poco frecuentes, la LLA-T exhibe proteínas de fusión de ABL1 con otros genes compañeros (por ejemplo, ETV6, BCR y EML1). Los inhibidores de tirosina–cinasas de ABL, como el imatinib o el dasatinib, quizás produzcan beneficios terapéuticos en este subtipo de LLA-T, pero la experiencia clínica con esta estrategia es muy limitada.
- Se notificaron fusiones génicas que afectan el gen SPI1 (codificador del factor de transcripción PU.1) en el 4 % de los niños japoneses con LLA-T. Los compañeros de fusión fueron STMN1 y TCF7. Los casos de LLA-T con fusiones de SPI1 tienen un pronóstico muy adverso; 6 de 7 personas afectadas murieron en el transcurso de 3 años desde el diagnóstico de una recaída temprana.
- El BCL11B es un factor de transcripción con dedos de cinc que cumple una función doble al activar y reprimir la transcripción. Se sabe que desempeña un papel importante en la diferenciación de las células T. En la LLA-T, el gen BCL11B participa en una translocación t(5;14)(q35;q32) donde un potenciador de BCL11B produce la expresión anómala de TLX3 (o NKX2-5). En el proceso de donación de su potenciador, se inactiva un alelo de BCL11B. Sin embargo, el estado haploinsuficiente resultante quizás también cumpla una función en la patogénesis del tumor. La función de BCL11B como gen supresor de tumores se apoya en los hallazgos que indican que cerca del 16 % de los pacientes tienen LLA-T que alberga deleciones o variantes de cambio de sentido. Como se describe en las secciones para leucemia linfoblástica aguda de células T precursoras tempranas y leucemia aguda de fenotipo mixto T/mieloide, en ocasiones BCL11B es leucemógeno por sobrexpresión.
- Otras fusiones génicas recurrentes en los pacientes con LLA-T son las que afectan los genes MLLT10, KMT2A, NUP214 y NUP98.
- Ploidía.
- Las anomalías recurrentes en el número de cromosomas son mucho menos frecuentes en la LLA-T que en la LLA-B. En un estudio se incluyeron 2250 pacientes pediátricos con LLA-T que se trataron en los protocolos de la Associazione Italiana di Ematologia e Oncologia Pediatrica y del Berlin-Frankfurt-Münster. En el estudio se encontró que la casi tetraploidía (índice de DNA, 1,79–2,28 o 81–103 cromosomas), presente en el 1,4 % de los pacientes, se relacionaba con características de la enfermedad y desenlaces favorables.
Características citogenéticas y genómicas de la leucemia linfoblástica aguda de células T precursoras tempranas
En la caracterización molecular detallada de la LLA de células T precursoras tempranas (TPT), se observó que esta entidad es muy heterogénea a nivel molecular, y no hay un solo gen afectado por una variante o alteración en el número de copias que se presente en más de un tercio de los casos. En comparación con otros casos de LLA-T, el grupo de TPT exhibió una tasa más baja de variantes de NOTCH1 y frecuencias significativamente más altas de alteraciones en los genes que regulan los receptores de citocinas y la señalización RAS, la maduración hematopoyética y las modificaciones en las histonas. El perfil transcripcional de la LLA TPT es parecido al de las células madre hematopoyéticas normales y las células madre de la leucemia mieloide.
En algunos estudios se encontró que la ausencia de la deleción bialélica del locus TCR-γ (ABD), detectado por hibridación genómica comparativa o DNA-PCR cuantitativa, se relacionó con un fracaso terapéutico temprano en los pacientes con LLA-T. ABD es característico de las células tímicas precursoras y muchos de los pacientes con LLA-T que exhiben ABD tienen un inmunofenotipo que coincide con el diagnóstico del fenotipo TPT.
La expresión aleloespecífica, por lo general alta, de BCL11B cumple una función oncogénica en un subgrupo de casos identificados como LLA TPT (7 de 58 casos en un estudio), así como en hasta de un 30 % a un 40 % de la leucemia aguda de fenotipo mixto T/M (LAFM T/M) de linaje ambiguo. La desregulación de la expresión de BCL11B puede ocurrir mediante múltiples mecanismos:
- Una de esas alteraciones es la t(2;14)(q22;q32), que produce una fusión génica ZEB2::BCL11B dentro del marco de lectura.
- Otras variantes estructurales que conducen a la desregulación de la expresión aleloespecífica de BCL11B incluyen variantes estructurales que unen secuencias reguladoras de genes activos (por ejemplo, ARID1B [cromosoma 6], BENC [cromosoma 7] y CDK6 [cromosoma 7]) secuencia arriba o secuencia abajo del locus de BCL11B lo que conduce a la expresión anómala en un proceso llamado secuestro de potenciador.
- Por último, en alrededor del 20 % de los casos con desregulación de la expresión de BCL11B, no se identifica ninguna translocación. En muchos de esos casos, la amplificación de un potenciador secuencia abajo, la amplificación en tándem del potenciador BCL11B (BETA), conduce a la transcripción impulsada por el promotor BCL11B.
- Hay una alta prevalencia de alteraciones de FLT3 y activación de JAK/STAT en las leucemias agudas causadas por alteraciones genómicas que conducen a la sobrexpresión de BCL11B.
Características citogenéticas y genómicas de la leucemia aguda de fenotipo mixto
El sistema de clasificación de la OMS para las leucemias de linaje ambiguo se resume en el Cuadro 3. Los criterios para la asignación del linaje durante el diagnóstico de la leucemia aguda de fenotipo mixto (LAFM) se describen en el Cuadro 4.
| Afección | Definición |
|---|---|
| LAFM = leucemia aguda de fenotipo mixto; SAI = sin otra indicación. | |
| aAdaptación de Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009. Obtenido del portal de Internet del Haematologica/the Hematology Journal http://www.haematologica.org. | |
| Leucemia aguda indiferenciada | Leucemia aguda que no expresa ningún marcador que se considere específico para el linaje linfoide ni el linaje mieloide |
| LAFM con BCR::ABL1 (t(9;22)(q34;q11.2)) | Leucemia aguda que cumple con los criterios diagnósticos de LAFM y se identifican blastocitos que también expresan la translocación (9;22) o el reordenamiento BCR::ABL1 |
| LAFM con KMT2A (t(v;11q23)) | Leucemia aguda que cumple con los criterios diagnósticos de la LAFM y se identifican blastocitos que también expresan una translocación que afecta el gen KMT2A |
| LAFM, B o mieloide, SAI (LAFM B/M) | Leucemia aguda que cumple con los criterios diagnósticos para asignar un linaje B y un linaje mieloide, y se identifican blastocitos que carecen de anomalías genéticas que afecten BCR::ABL1 o KMT2A |
| LAFM, T o mieloide, SAI (LAFM T/M) | Leucemia aguda que cumple con los criterios diagnósticos para asignar un linaje T y un linaje mieloide, y se identifican blastocitos que carecen de anomalías genéticas que afectan BCR::ABL1 o KMT2A |
| LAFM, B o mieloide, SAI (tipos poco frecuentes) | Leucemia aguda que cumple con los criterios diagnósticos para asignar un linaje B y un linaje T |
| Otras leucemias de linaje ambiguo | Leucemia o linfoma linfoblásticos de células citolíticas naturales |
| Linaje | Criterios |
|---|---|
| aAdaptado de Arber et al. | |
| bFuerte se define como más brillante o igual a las células B o T normales de la muestra. | |
| Linaje mieloide | Mieloperoxidasa (citometría de flujo, prueba inmunohistoquímica o citoquímica); o diferenciación monocítica (por lo menos dos de los siguientes aspectos: prueba citoquímica de esterasa inespecífica, CD11c, CD14, CD64 o lisozima) |
| Linaje T | CD3 citoplasmático fuerteb (con anticuerpos contra la cadena ε de CD3); o CD3 de superficie |
| Linaje B | CD19 con expresión fuerteb de por lo menos una de las siguientes moléculas: CD79a, CD22 citoplasmático o CD10; o CD19 débil y por lo menos expresión fuerte de dos de las siguientes moléculas: CD79a, CD22 citoplasmático o CD10 |
El sistema de clasificación de la LAFM incluye dos entidades definidas a partir de la alteración molecular principal: LAFM con translocación BCR::ABL1 y LAFM con reordenamiento de KMT2A. Las alteraciones genómicas asociadas con cada una de las entidades LAFM, B/mieloide, SAI (LAFM B/M) y LAFM, T/mieloide, SAI (LAFM T/M) son diferentes, como se describe a continuación:
- LAFM B/M.
- Entre 115 casos de LAFM en los que se hizo la caracterización genómica, se encontraron 35 (30 %) casos de LAFM B/M. Además hubo 16 casos de LAFM (14 %) con reordenamientos de KMT2A, 15 de ellos exhibían un inmunofenotipo B/mieloide.
- Alrededor de la mitad de los casos de LAFM B/M tenían reordenamientos de ZNF384 con compañeros de fusión recurrentes, como TCF3 y EP300. Estos casos exhibieron perfiles de expresión génica indistinguibles de los casos de LLA-B con reordenamientos de ZNF384.
- Cerca de dos tercios de los casos de LAFM B/M presentaban alteraciones en la vía RAS, los genes alterados de manera más frecuente fueron NRAS y PTPN11.
- Los genes codificadores de reguladores epigenéticos (por ejemplo, MLLT3, KDM6A, EP300 y CREBBP) están alterados en alrededor de dos tercios de los casos de LAFM B/M.
- LAFM T/M.
- Entre 115 casos de LAFM en los que se hizo la caracterización genómica, se encontraron 49 (43 %) casos de LAFM T/M. Las características genómicas de los casos de LAFM T/M comparten semejanzas con la LLA TPT lo que indica que la LAFM T/M y la LLA TPT son entidades similares a lo largo de la variedad de leucemias inmaduras.
- En comparación con la LLA-T, la LAFM T/M presentó una tasa más baja de alteraciones en los factores de transcripción centrales de la LLA-T (TAL1, TAL2, TLX1, TLX3, LMO1, LMO2, NKX2-1, HOXA10 y LYL1) (63 vs. 16 %, respectivamente). También se observó una tasa baja similar en la LLA TPT.
- Las variantes de CDKN2A, CDKN2B y NOTCH1, presentes en alrededor de dos tercios de los casos de LLA-T, son mucho menos comunes en la LAFM T/M. Por el contrario, las variantes de WT1 se presentan en alrededor del 40 % de los casos de LAFM T/M, pero en menos del 10 % de los casos de LLA-T.
- Un tercio de los casos de LAFM T/M tienen alteraciones genómicas relacionadas con BCL11B que producen la expresión aleloespecífica, por lo general elevada, de BCL11B.
- Una de esas alteraciones es la t(2;14)(q22;q32), que produce una fusión génica ZEB2::BCL11B dentro del marco de lectura que conduce a la desregulación de la expresión de BCL11B.
- Otras variantes estructurales que conducen a la desregulación de la expresión aleloespecífica de BCL11B incluyen variantes estructurales que unen secuencias reguladoras de genes activos (por ejemplo, ARID1B [cromosoma 6], BENC [cromosoma 7] y CDK6 [cromosoma 7]) secuencia arriba o secuencia abajo del locus de BCL11B en un proceso llamado secuestro de potenciador.
- Por último, en alrededor del 20 % de los casos con desregulación de la expresión de BCL11B no se identifica ninguna translocación. En estos casos, la amplificación de un potenciador secuencia abajo, la amplificación en tándem del potenciador BCL11B (BETA), conduce a la transcripción impulsada por el promotor BCL11B.
- Hay una alta prevalencia de alteraciones de FLT3 y activación de JAK/STAT en las leucemias agudas causadas por alteraciones genómicas que conducen a la sobrexpresión de BCL11B.
- Las variantes de las vías RAS y JAK-STAT son comunes en los casos de LAFM T/M y LLA TPT, mientras que las alteraciones en la vía de señalización PI3K son más comunes en la LLA-T. En la LAFM T/M, el gen de vía de señalización alterado de manera más frecuente fue FLT3 (43 % de los casos). Las variantes de FLT3 tienden a ser mutuamente excluyentes de las variantes de la vía RAS.
- Los genes codificadores de reguladores epigenéticos (por ejemplo, EZH2 y PHF6) están alterados en alrededor de dos tercios de los casos de LAFM T/M.
Polimorfismos génicos en las vías metabólicas de los fármacos
Se ha notificado que varios polimorfismos de los genes involucrados en el metabolismo de fármacos quimioterapéuticos tienen importancia pronóstica en la LLA infantil.
- TPMT.
Los pacientes con fenotipos de variantes de TPMT (gen que participa en el metabolismo de las tiopurinas, como la mercaptopurina) tienen desenlaces más favorables, aunque estos pacientes también exhiben un riesgo más alto de presentar importantes efectos tóxicos relacionados con el tratamiento, como mielodepresión, infección y segundas neoplasias malignas. Los pacientes con homocigosis para variantes de TPMT relacionadas con actividad enzimática baja solo toleran dosis muy bajas de mercaptopurina (alrededor del 10 % de la dosis estándar) y se tratan con dosis reducidas de este medicamento para evitar toxicidad excesiva. Los pacientes heterocigóticos para la variante de este gen de enzima por lo general toleran la mercaptopurina sin toxicidad grave, pero necesitan reducciones de dosis más frecuentes debido a toxicidad hematopoyética que los pacientes homocigóticos para el alelo normal.
- NUDT15.
Las variantes de la línea germinal en NUDT15 que reducen o anulan la actividad de esta enzima también disminuyen la tolerabilidad a las tiopurinas. Las variantes de NUDT15 son más comunes en los pacientes del este de Asía y en los pacientes hispanos, pero son infrecuentes en los pacientes europeos y africanos. Los pacientes homocigóticos para las variantes de riesgo toleran solo dosis muy bajas de mercaptopurina, mientras que los pacientes heterocigóticos para los alelos de riesgo toleran dosis más bajas que los pacientes homocigóticos para el alelo de tipo natural (reducción promedio de la dosis, 25 %), pero hay una superposición amplia de las dosis toleradas entre los dos grupos.
- CEP72.
Los polimorfismos génicos también afectan la expresión de las proteínas que cumplen funciones importantes en los efectos celulares de los antineoplásicos. Por ejemplo, los pacientes homocigóticos para un polimorfismo en la región promotora de CEP72 (una proteína del centrosoma que participa en la formación de microtúbulos) tienen un riesgo aumentado de neurotoxicidad por vincristina.
- Polimorfismos de un solo nucleótido.
En el análisis de polimorfismos en el genoma completo, se han identificado polimorfismos de un solo nucleótido específicos que se relacionan con una ERM alta al final de la inducción y riesgo de recaída. Los polimorfismos de la interleucina-15, y los genes asociados con el metabolismo del etopósido y el metotrexato, exhibieron una asociación significativa con la respuesta al tratamiento en dos cohortes numerosas de pacientes con LLA tratados con protocolos del SJCRH y del COG. Las variantes polimórficas que afectan el transportador de folato reducido y el metabolismo del metotrexato se relacionaron con la toxicidad y el desenlace. Aunque estas asociaciones indican que las variaciones individuales en el metabolismo de los fármacos quizás afecten el desenlace, se han realizado pocos estudios para intentar ajustar a partir de dichas variaciones; se desconoce si una modificación personalizada de la dosis a partir de estos hallazgos mejoraría los resultados.
References
- Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet 54 (9): 1376-1389, 2022.
- Mullighan CG, Goorha S, Radtke I, et al.: Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446 (7137): 758-64, 2007.
- Mullighan CG, Miller CB, Radtke I, et al.: BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453 (7191): 110-4, 2008.
- Roberts KG, Li Y, Payne-Turner D, et al.: Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 371 (11): 1005-15, 2014.
- Lilljebjörn H, Henningsson R, Hyrenius-Wittsten A, et al.: Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun 7: 11790, 2016.
- Zhang J, McCastlain K, Yoshihara H, et al.: Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet 48 (12): 1481-1489, 2016.
- Holmfeldt L, Wei L, Diaz-Flores E, et al.: The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45 (3): 242-52, 2013.
- Loh ML, Zhang J, Harvey RC, et al.: Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children's Oncology Group TARGET Project. Blood 121 (3): 485-8, 2013.
- Bercovich D, Ganmore I, Scott LM, et al.: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484-92, 2008.
- Li Z, Chang TC, Junco JJ, et al.: Genomic landscape of Down syndrome-associated acute lymphoblastic leukemia. Blood 142 (2): 172-184, 2023.
- Andersson AK, Ma J, Wang J, et al.: The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet 47 (4): 330-7, 2015.
- Ma X, Edmonson M, Yergeau D, et al.: Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun 6: 6604, 2015.
- Meyer JA, Wang J, Hogan LE, et al.: Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 45 (3): 290-4, 2013.
- Li B, Li H, Bai Y, et al.: Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med 21 (6): 563-71, 2015.
- Mullighan CG, Zhang J, Kasper LH, et al.: CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471 (7337): 235-9, 2011.
- Mattano LA, Devidas M, Maloney KW, et al.: Favorable Trisomies and ETV6-RUNX1 Predict Cure in Low-Risk B-Cell Acute Lymphoblastic Leukemia: Results From Children's Oncology Group Trial AALL0331. J Clin Oncol 39 (14): 1540-1552, 2021.
- Moorman AV, Ensor HM, Richards SM, et al.: Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol 11 (5): 429-38, 2010.
- Alaggio R, Amador C, Anagnostopoulos I, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 36 (7): 1720-1748, 2022.
- Paulsson K, Johansson B: High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 48 (8): 637-60, 2009.
- Aricò M, Valsecchi MG, Rizzari C, et al.: Long-term results of the AIEOP-ALL-95 Trial for Childhood Acute Lymphoblastic Leukemia: insight on the prognostic value of DNA index in the framework of Berlin-Frankfurt-Muenster based chemotherapy. J Clin Oncol 26 (2): 283-9, 2008.
- Dastugue N, Suciu S, Plat G, et al.: Hyperdiploidy with 58-66 chromosomes in childhood B-acute lymphoblastic leukemia is highly curable: 58951 CLG-EORTC results. Blood 121 (13): 2415-23, 2013.
- Synold TW, Relling MV, Boyett JM, et al.: Blast cell methotrexate-polyglutamate accumulation in vivo differs by lineage, ploidy, and methotrexate dose in acute lymphoblastic leukemia. J Clin Invest 94 (5): 1996-2001, 1994.
- Moorman AV, Richards SM, Martineau M, et al.: Outcome heterogeneity in childhood high-hyperdiploid acute lymphoblastic leukemia. Blood 102 (8): 2756-62, 2003.
- Chilton L, Buck G, Harrison CJ, et al.: High hyperdiploidy among adolescents and adults with acute lymphoblastic leukaemia (ALL): cytogenetic features, clinical characteristics and outcome. Leukemia 28 (7): 1511-8, 2014.
- Sutcliffe MJ, Shuster JJ, Sather HN, et al.: High concordance from independent studies by the Children's Cancer Group (CCG) and Pediatric Oncology Group (POG) associating favorable prognosis with combined trisomies 4, 10, and 17 in children with NCI Standard-Risk B-precursor Acute Lymphoblastic Leukemia: a Children's Oncology Group (COG) initiative. Leukemia 19 (5): 734-40, 2005.
- Harris MB, Shuster JJ, Carroll A, et al.: Trisomy of leukemic cell chromosomes 4 and 10 identifies children with B-progenitor cell acute lymphoblastic leukemia with a very low risk of treatment failure: a Pediatric Oncology Group study. Blood 79 (12): 3316-24, 1992.
- Enshaei A, Vora A, Harrison CJ, et al.: Defining low-risk high hyperdiploidy in patients with paediatric acute lymphoblastic leukaemia: a retrospective analysis of data from the UKALL97/99 and UKALL2003 clinical trials. Lancet Haematol 8 (11): e828-e839, 2021.
- Heerema NA, Harbott J, Galimberti S, et al.: Secondary cytogenetic aberrations in childhood Philadelphia chromosome positive acute lymphoblastic leukemia are nonrandom and may be associated with outcome. Leukemia 18 (4): 693-702, 2004.
- Carroll AJ, Shago M, Mikhail FM, et al.: Masked hypodiploidy: Hypodiploid acute lymphoblastic leukemia (ALL) mimicking hyperdiploid ALL in children: A report from the Children's Oncology Group. Cancer Genet 238: 62-68, 2019.
- Nachman JB, Heerema NA, Sather H, et al.: Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood 110 (4): 1112-5, 2007.
- Raimondi SC, Zhou Y, Shurtleff SA, et al.: Near-triploidy and near-tetraploidy in childhood acute lymphoblastic leukemia: association with B-lineage blast cells carrying the ETV6-RUNX1 fusion, T-lineage immunophenotype, and favorable outcome. Cancer Genet Cytogenet 169 (1): 50-7, 2006.
- Attarbaschi A, Mann G, König M, et al.: Incidence and relevance of secondary chromosome abnormalities in childhood TEL/AML1+ acute lymphoblastic leukemia: an interphase FISH analysis. Leukemia 18 (10): 1611-6, 2004.
- Lemez P, Attarbaschi A, Béné MC, et al.: Childhood near-tetraploid acute lymphoblastic leukemia: an EGIL study on 36 cases. Eur J Haematol 85 (4): 300-8, 2010.
- Paulsson K, Lilljebjörn H, Biloglav A, et al.: The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet 47 (6): 672-6, 2015.
- Harrison CJ, Moorman AV, Broadfield ZJ, et al.: Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol 125 (5): 552-9, 2004.
- Mullighan CG, Jeha S, Pei D, et al.: Outcome of children with hypodiploid ALL treated with risk-directed therapy based on MRD levels. Blood 126 (26): 2896-9, 2015.
- Pui CH, Rebora P, Schrappe M, et al.: Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Retrospective Multinational Study. J Clin Oncol 37 (10): 770-779, 2019.
- McNeer JL, Devidas M, Dai Y, et al.: Hematopoietic Stem-Cell Transplantation Does Not Improve the Poor Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group. J Clin Oncol 37 (10): 780-789, 2019.
- Irving J, Matheson E, Minto L, et al.: Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 124 (23): 3420-30, 2014.
- Qian M, Cao X, Devidas M, et al.: TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J Clin Oncol 36 (6): 591-599, 2018.
- Rubnitz JE, Wichlan D, Devidas M, et al.: Prospective analysis of TEL gene rearrangements in childhood acute lymphoblastic leukemia: a Children's Oncology Group study. J Clin Oncol 26 (13): 2186-91, 2008.
- Kanerva J, Saarinen-Pihkala UM, Niini T, et al.: Favorable outcome in 20-year follow-up of children with very-low-risk ALL and minimal standard therapy, with special reference to TEL-AML1 fusion. Pediatr Blood Cancer 42 (1): 30-5, 2004.
- Aldrich MC, Zhang L, Wiemels JL, et al.: Cytogenetics of Hispanic and White children with acute lymphoblastic leukemia in California. Cancer Epidemiol Biomarkers Prev 15 (3): 578-81, 2006.
- Loh ML, Goldwasser MA, Silverman LB, et al.: Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood 107 (11): 4508-13, 2006.
- Borowitz MJ, Devidas M, Hunger SP, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood 111 (12): 5477-85, 2008.
- Madzo J, Zuna J, Muzíková K, et al.: Slower molecular response to treatment predicts poor outcome in patients with TEL/AML1 positive acute lymphoblastic leukemia: prospective real-time quantitative reverse transcriptase-polymerase chain reaction study. Cancer 97 (1): 105-13, 2003.
- Bhojwani D, Pei D, Sandlund JT, et al.: ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy. Leukemia 26 (2): 265-70, 2012.
- Enshaei A, Schwab CJ, Konn ZJ, et al.: Long-term follow-up of ETV6-RUNX1 ALL reveals that NCI risk, rather than secondary genetic abnormalities, is the key risk factor. Leukemia 27 (11): 2256-9, 2013.
- Barbany G, Andersen MK, Autio K, et al.: Additional aberrations of the ETV6 and RUNX1 genes have no prognostic impact in 229 t(12;21)(p13;q22)-positive B-cell precursor acute lymphoblastic leukaemias treated according to the NOPHO-ALL-2000 protocol. Leuk Res 36 (7): 936-8, 2012.
- Forestier E, Heyman M, Andersen MK, et al.: Outcome of ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia in the NOPHO-ALL-1992 protocol: frequent late relapses but good overall survival. Br J Haematol 140 (6): 665-72, 2008.
- Seeger K, Stackelberg AV, Taube T, et al.: Relapse of TEL-AML1--positive acute lymphoblastic leukemia in childhood: a matched-pair analysis. J Clin Oncol 19 (13): 3188-93, 2001.
- Gandemer V, Chevret S, Petit A, et al.: Excellent prognosis of late relapses of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia: lessons from the FRALLE 93 protocol. Haematologica 97 (11): 1743-50, 2012.
- Zuna J, Ford AM, Peham M, et al.: TEL deletion analysis supports a novel view of relapse in childhood acute lymphoblastic leukemia. Clin Cancer Res 10 (16): 5355-60, 2004.
- van Delft FW, Horsley S, Colman S, et al.: Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 117 (23): 6247-54, 2011.
- Aricò M, Schrappe M, Hunger SP, et al.: Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol 28 (31): 4755-61, 2010.
- Schrappe M, Aricò M, Harbott J, et al.: Philadelphia chromosome-positive (Ph+) childhood acute lymphoblastic leukemia: good initial steroid response allows early prediction of a favorable treatment outcome. Blood 92 (8): 2730-41, 1998.
- Ribeiro RC, Broniscer A, Rivera GK, et al.: Philadelphia chromosome-positive acute lymphoblastic leukemia in children: durable responses to chemotherapy associated with low initial white blood cell counts. Leukemia 11 (9): 1493-6, 1997.
- Biondi A, Schrappe M, De Lorenzo P, et al.: Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol 13 (9): 936-45, 2012.
- Schultz KR, Bowman WP, Aledo A, et al.: Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 27 (31): 5175-81, 2009.
- Schultz KR, Carroll A, Heerema NA, et al.: Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's Oncology Group study AALL0031. Leukemia 28 (7): 1467-71, 2014.
- Duffield AS, Mullighan CG, Borowitz MJ: International Consensus Classification of acute lymphoblastic leukemia/lymphoma. Virchows Arch 482 (1): 11-26, 2023.
- Short NJ, Jabbour E, Macaron W, et al.: Ultrasensitive NGS MRD assessment in Ph+ ALL: Prognostic impact and correlation with RT-PCR for BCR::ABL1. Am J Hematol 98 (8): 1196-1203, 2023.
- Hovorkova L, Zaliova M, Venn NC, et al.: Monitoring of childhood ALL using BCR-ABL1 genomic breakpoints identifies a subgroup with CML-like biology. Blood 129 (20): 2771-2781, 2017.
- Zuna J, Hovorkova L, Krotka J, et al.: Minimal residual disease in BCR::ABL1-positive acute lymphoblastic leukemia: different significance in typical ALL and in CML-like disease. Leukemia 36 (12): 2793-2801, 2022.
- Hunger SP, Tran TH, Saha V, et al.: Dasatinib with intensive chemotherapy in de novo paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (CA180-372/COG AALL1122): a single-arm, multicentre, phase 2 trial. Lancet Haematol 10 (7): e510-e520, 2023.
- Pui CH, Chessells JM, Camitta B, et al.: Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia 17 (4): 700-6, 2003.
- Johansson B, Moorman AV, Haas OA, et al.: Hematologic malignancies with t(4;11)(q21;q23)--a cytogenetic, morphologic, immunophenotypic and clinical study of 183 cases. European 11q23 Workshop participants. Leukemia 12 (5): 779-87, 1998.
- Raimondi SC, Peiper SC, Kitchingman GR, et al.: Childhood acute lymphoblastic leukemia with chromosomal breakpoints at 11q23. Blood 73 (6): 1627-34, 1989.
- Harrison CJ, Moorman AV, Barber KE, et al.: Interphase molecular cytogenetic screening for chromosomal abnormalities of prognostic significance in childhood acute lymphoblastic leukaemia: a UK Cancer Cytogenetics Group Study. Br J Haematol 129 (4): 520-30, 2005.
- Pui CH, Pei D, Campana D, et al.: A revised definition for cure of childhood acute lymphoblastic leukemia. Leukemia 28 (12): 2336-43, 2014.
- Pieters R, Schrappe M, De Lorenzo P, et al.: A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370 (9583): 240-50, 2007.
- Attarbaschi A, Möricke A, Harrison CJ, et al.: Outcomes of Childhood Noninfant Acute Lymphoblastic Leukemia With 11q23/KMT2A Rearrangements in a Modern Therapy Era: A Retrospective International Study. J Clin Oncol 41 (7): 1404-1422, 2023.
- Isobe T, Takagi M, Sato-Otsubo A, et al.: Multi-omics analysis defines highly refractory RAS burdened immature subgroup of infant acute lymphoblastic leukemia. Nat Commun 13 (1): 4501, 2022.
- Pui CH, Gaynon PS, Boyett JM, et al.: Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet 359 (9321): 1909-15, 2002.
- Rubnitz JE, Camitta BM, Mahmoud H, et al.: Childhood acute lymphoblastic leukemia with the MLL-ENL fusion and t(11;19)(q23;p13.3) translocation. J Clin Oncol 17 (1): 191-6, 1999.
- Hunger SP: Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood 87 (4): 1211-24, 1996.
- Uckun FM, Sensel MG, Sather HN, et al.: Clinical significance of translocation t(1;19) in childhood acute lymphoblastic leukemia in the context of contemporary therapies: a report from the Children's Cancer Group. J Clin Oncol 16 (2): 527-35, 1998.
- Fischer U, Forster M, Rinaldi A, et al.: Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet 47 (9): 1020-9, 2015.
- Pui CH, Sandlund JT, Pei D, et al.: Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA 290 (15): 2001-7, 2003.
- Crist WM, Carroll AJ, Shuster JJ, et al.: Poor prognosis of children with pre-B acute lymphoblastic leukemia is associated with the t(1;19)(q23;p13): a Pediatric Oncology Group study. Blood 76 (1): 117-22, 1990.
- Andersen MK, Autio K, Barbany G, et al.: Paediatric B-cell precursor acute lymphoblastic leukaemia with t(1;19)(q23;p13): clinical and cytogenetic characteristics of 47 cases from the Nordic countries treated according to NOPHO protocols. Br J Haematol 155 (2): 235-43, 2011.
- Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.
- Jeha S, Pei D, Raimondi SC, et al.: Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia 23 (8): 1406-9, 2009.
- Minson KA, Prasad P, Vear S, et al.: t(17;19) in Children with Acute Lymphocytic Leukemia: A Report of 3 Cases and a Review of the Literature. Case Rep Hematol 2013: 563291, 2013.
- Lee SHR, Antillon-Klussmann F, Pei D, et al.: Association of Genetic Ancestry With the Molecular Subtypes and Prognosis of Childhood Acute Lymphoblastic Leukemia. JAMA Oncol 8 (3): 354-363, 2022.
- Zaliova M, Potuckova E, Hovorkova L, et al.: ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on screening method sensitivity. Haematologica 104 (7): 1407-1416, 2019.
- Harvey RC, Mullighan CG, Wang X, et al.: Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 116 (23): 4874-84, 2010.
- Zaliova M, Zimmermannova O, Dörge P, et al.: ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia 28 (1): 182-5, 2014.
- Clappier E, Auclerc MF, Rapion J, et al.: An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia 28 (1): 70-7, 2014.
- Li Z, Lee SHR, Chin WHN, et al.: Distinct clinical characteristics of DUX4- and PAX5-altered childhood B-lymphoblastic leukemia. Blood Adv 5 (23): 5226-5238, 2021.
- Gu Z, Churchman M, Roberts K, et al.: Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun 7: 13331, 2016.
- Liu YF, Wang BY, Zhang WN, et al.: Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 8: 173-83, 2016.
- Suzuki K, Okuno Y, Kawashima N, et al.: MEF2D-BCL9 Fusion Gene Is Associated With High-Risk Acute B-Cell Precursor Lymphoblastic Leukemia in Adolescents. J Clin Oncol 34 (28): 3451-9, 2016.
- Lilljebjörn H, Ågerstam H, Orsmark-Pietras C, et al.: RNA-seq identifies clinically relevant fusion genes in leukemia including a novel MEF2D/CSF1R fusion responsive to imatinib. Leukemia 28 (4): 977-9, 2014.
- Hirabayashi S, Ohki K, Nakabayashi K, et al.: ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 102 (1): 118-129, 2017.
- Qian M, Zhang H, Kham SK, et al.: Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res 27 (2): 185-195, 2017.
- Hirabayashi S, Butler ER, Ohki K, et al.: Clinical characteristics and outcomes of B-ALL with ZNF384 rearrangements: a retrospective analysis by the Ponte di Legno Childhood ALL Working Group. Leukemia 35 (11): 3272-3277, 2021.
- Shago M, Abla O, Hitzler J, et al.: Frequency and outcome of pediatric acute lymphoblastic leukemia with ZNF384 gene rearrangements including a novel translocation resulting in an ARID1B/ZNF384 gene fusion. Pediatr Blood Cancer 63 (11): 1915-21, 2016.
- Yao L, Cen J, Pan J, et al.: TAF15-ZNF384 fusion gene in childhood mixed phenotype acute leukemia. Cancer Genet 211: 1-4, 2017.
- Alexander TB, Gu Z, Iacobucci I, et al.: The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 562 (7727): 373-379, 2018.
- Boer JM, Valsecchi MG, Hormann FM, et al.: Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia 35 (10): 2978-2982, 2021.
- De Lorenzo P, Moorman AV, Pieters R, et al.: Cytogenetics and outcome of infants with acute lymphoblastic leukemia and absence of MLL rearrangements. Leukemia 28 (2): 428-30, 2014.
- Fazio G, Bardini M, De Lorenzo P, et al.: Recurrent genetic fusions redefine MLL germ line acute lymphoblastic leukemia in infants. Blood 137 (14): 1980-1984, 2021.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Hogan TF, Koss W, Murgo AJ, et al.: Acute lymphoblastic leukemia with chromosomal 5;14 translocation and hypereosinophilia: case report and literature review. J Clin Oncol 5 (3): 382-90, 1987.
- Grimaldi JC, Meeker TC: The t(5;14) chromosomal translocation in a case of acute lymphocytic leukemia joins the interleukin-3 gene to the immunoglobulin heavy chain gene. Blood 73 (8): 2081-5, 1989.
- Meeker TC, Hardy D, Willman C, et al.: Activation of the interleukin-3 gene by chromosome translocation in acute lymphocytic leukemia with eosinophilia. Blood 76 (2): 285-9, 1990.
- Sutton R, Lonergan M, Tapp H, et al.: Two cases of hypereosinophilia and high-risk acute lymphoblastic leukemia. Leukemia 22 (7): 1463-5, 2008.
- Koleilat A, Smadbeck JB, Zepeda-Mendoza CJ, et al.: Characterization of unusual iAMP21 B-lymphoblastic leukemia (iAMP21-ALL) from the Mayo Clinic and Children's Oncology Group. Genes Chromosomes Cancer 61 (12): 710-719, 2022.
- Heerema NA, Carroll AJ, Devidas M, et al.: Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk children's oncology group studies: a report from the children's oncology group. J Clin Oncol 31 (27): 3397-402, 2013.
- Moorman AV, Robinson H, Schwab C, et al.: Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: a comparison of the MRC ALL97/99 and UKALL2003 trials. J Clin Oncol 31 (27): 3389-96, 2013.
- Harrison CJ, Moorman AV, Schwab C, et al.: An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia 28 (5): 1015-21, 2014.
- Gu Z, Churchman ML, Roberts KG, et al.: PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet 51 (2): 296-307, 2019.
- Strehl S, König M, Dworzak MN, et al.: PAX5/ETV6 fusion defines cytogenetic entity dic(9;12)(p13;p13). Leukemia 17 (6): 1121-3, 2003.
- Schwab C, Nebral K, Chilton L, et al.: Intragenic amplification of PAX5: a novel subgroup in B-cell precursor acute lymphoblastic leukemia? Blood Adv 1 (19): 1473-7, 2017.
- Den Boer ML, van Slegtenhorst M, De Menezes RX, et al.: A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 10 (2): 125-34, 2009.
- Mullighan CG, Su X, Zhang J, et al.: Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 360 (5): 470-80, 2009.
- Reshmi SC, Harvey RC, Roberts KG, et al.: Targetable kinase gene fusions in high-risk B-ALL: a study from the Children's Oncology Group. Blood 129 (25): 3352-3361, 2017.
- Roberts KG, Morin RD, Zhang J, et al.: Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 22 (2): 153-66, 2012.
- van der Veer A, Waanders E, Pieters R, et al.: Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood 122 (15): 2622-9, 2013.
- Roberts KG, Reshmi SC, Harvey RC, et al.: Genomic and outcome analyses of Ph-like ALL in NCI standard-risk patients: a report from the Children's Oncology Group. Blood 132 (8): 815-824, 2018.
- Roberts KG, Pei D, Campana D, et al.: Outcomes of children with BCR-ABL1–like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol 32 (27): 3012-20, 2014.
- Harvey RC, Mullighan CG, Chen IM, et al.: Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 115 (26): 5312-21, 2010.
- Mullighan CG, Collins-Underwood JR, Phillips LA, et al.: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243-6, 2009.
- Schwab C, Roberts K, Boer JM, et al.: SSBP2-CSF1R is a recurrent fusion in B-lineage acute lymphoblastic leukemia with diverse genetic presentation and variable outcome. Blood 137 (13): 1835-1838, 2021.
- den Boer ML, Cario G, Moorman AV, et al.: Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: a multicentre, retrospective, cohort study. Lancet Haematol 8 (1): e55-e66, 2021.
- Iacobucci I, Li Y, Roberts KG, et al.: Truncating Erythropoietin Receptor Rearrangements in Acute Lymphoblastic Leukemia. Cancer Cell 29 (2): 186-200, 2016.
- Cario G, Zimmermann M, Romey R, et al.: Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood 115 (26): 5393-7, 2010.
- Ensor HM, Schwab C, Russell LJ, et al.: Demographic, clinical, and outcome features of children with acute lymphoblastic leukemia and CRLF2 deregulation: results from the MRC ALL97 clinical trial. Blood 117 (7): 2129-36, 2011.
- Schmäh J, Fedders B, Panzer-Grümayer R, et al.: Molecular characterization of acute lymphoblastic leukemia with high CRLF2 gene expression in childhood. Pediatr Blood Cancer 64 (10): , 2017.
- Raca G, Abdel-Azim H, Yue F, et al.: Increased Incidence of IKZF1 deletions and IGH-CRLF2 translocations in B-ALL of Hispanic/Latino children-a novel health disparity. Leukemia 35 (8): 2399-2402, 2021.
- Vesely C, Frech C, Eckert C, et al.: Genomic and transcriptional landscape of P2RY8-CRLF2-positive childhood acute lymphoblastic leukemia. Leukemia 31 (7): 1491-1501, 2017.
- Russell LJ, Jones L, Enshaei A, et al.: Characterisation of the genomic landscape of CRLF2-rearranged acute lymphoblastic leukemia. Genes Chromosomes Cancer 56 (5): 363-372, 2017.
- Potter N, Jones L, Blair H, et al.: Single-cell analysis identifies CRLF2 rearrangements as both early and late events in Down syndrome and non-Down syndrome acute lymphoblastic leukaemia. Leukemia 33 (4): 893-904, 2019.
- Morak M, Attarbaschi A, Fischer S, et al.: Small sizes and indolent evolutionary dynamics challenge the potential role of P2RY8-CRLF2-harboring clones as main relapse-driving force in childhood ALL. Blood 120 (26): 5134-42, 2012.
- Schwab CJ, Chilton L, Morrison H, et al.: Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica 98 (7): 1081-8, 2013.
- Chen IM, Harvey RC, Mullighan CG, et al.: Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 119 (15): 3512-22, 2012.
- Palmi C, Vendramini E, Silvestri D, et al.: Poor prognosis for P2RY8-CRLF2 fusion but not for CRLF2 over-expression in children with intermediate risk B-cell precursor acute lymphoblastic leukemia. Leukemia 26 (10): 2245-53, 2012.
- Clappier E, Grardel N, Bakkus M, et al.: IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children's Leukemia Group study 58951. Leukemia 29 (11): 2154-61, 2015.
- Srinivasan S, Ramanathan S, Kumar S, et al.: Prevalence and prognostic significance of IKZF1 deletion in paediatric acute lymphoblastic leukemia: A systematic review and meta-analysis. Ann Hematol 102 (8): 2165-2179, 2023.
- Buitenkamp TD, Pieters R, Gallimore NE, et al.: Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia 26 (10): 2204-11, 2012.
- Krentz S, Hof J, Mendioroz A, et al.: Prognostic value of genetic alterations in children with first bone marrow relapse of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia 27 (2): 295-304, 2013.
- Feng J, Tang Y: Prognostic significance of IKZF1 alteration status in pediatric B-lineage acute lymphoblastic leukemia: a meta-analysis. Leuk Lymphoma 54 (4): 889-91, 2013.
- Dörge P, Meissner B, Zimmermann M, et al.: IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica 98 (3): 428-32, 2013.
- Olsson L, Castor A, Behrendtz M, et al.: Deletions of IKZF1 and SPRED1 are associated with poor prognosis in a population-based series of pediatric B-cell precursor acute lymphoblastic leukemia diagnosed between 1992 and 2011. Leukemia 28 (2): 302-10, 2014.
- Boer JM, van der Veer A, Rizopoulos D, et al.: Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia 30 (1): 32-8, 2016.
- Tran TH, Harris MH, Nguyen JV, et al.: Prognostic impact of kinase-activating fusions and IKZF1 deletions in pediatric high-risk B-lineage acute lymphoblastic leukemia. Blood Adv 2 (5): 529-533, 2018.
- Vrooman LM, Blonquist TM, Harris MH, et al.: Refining risk classification in childhood B acute lymphoblastic leukemia: results of DFCI ALL Consortium Protocol 05-001. Blood Adv 2 (12): 1449-1458, 2018.
- Öfverholm I, Rezayee F, Heyman M, et al.: The prognostic impact of IKZF1 deletions and UKALL genetic classifiers in paediatric B-cell precursor acute lymphoblastic leukaemia treated according to NOPHO 2008 protocols. Br J Haematol 202 (2): 384-392, 2023.
- van der Veer A, Zaliova M, Mottadelli F, et al.: IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood 123 (11): 1691-8, 2014.
- Stanulla M, Dagdan E, Zaliova M, et al.: IKZF1plus Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J Clin Oncol 36 (12): 1240-1249, 2018.
- Mangum DS, Meyer JA, Mason CC, et al.: Association of Combined Focal 22q11.22 Deletion and IKZF1 Alterations With Outcomes in Childhood Acute Lymphoblastic Leukemia. JAMA Oncol 7 (10): 1521-1528, 2021.
- Yeoh AEJ, Lu Y, Chin WHN, et al.: Intensifying Treatment of Childhood B-Lymphoblastic Leukemia With IKZF1 Deletion Reduces Relapse and Improves Overall Survival: Results of Malaysia-Singapore ALL 2010 Study. J Clin Oncol 36 (26): 2726-2735, 2018.
- Pieters R, de Groot-Kruseman H, Fiocco M, et al.: Improved Outcome for ALL by Prolonging Therapy for IKZF1 Deletion and Decreasing Therapy for Other Risk Groups. J Clin Oncol 41 (25): 4130-4142, 2023.
- Herbrueggen H, Mueller S, Rohde J, et al.: Treatment and outcome of IG-MYC+ neoplasms with precursor B-cell phenotype in childhood and adolescence. Leukemia 34 (3): 942-946, 2020.
- Sakaguchi K, Imamura T, Ishimaru S, et al.: Nationwide study of pediatric B-cell precursor acute lymphoblastic leukemia with chromosome 8q24/MYC rearrangement in Japan. Pediatr Blood Cancer 67 (7): e28341, 2020.
- Bomken S, Enshaei A, Schwalbe EC, et al.: Molecular characterization and clinical outcome of B-cell precursor acute lymphoblastic leukemia with IG-MYC rearrangement. Haematologica 108 (3): 717-731, 2023.
- Liu Y, Easton J, Shao Y, et al.: The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 49 (8): 1211-1218, 2017.
- Armstrong SA, Look AT: Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol 23 (26): 6306-15, 2005.
- Karrman K, Forestier E, Heyman M, et al.: Clinical and cytogenetic features of a population-based consecutive series of 285 pediatric T-cell acute lymphoblastic leukemias: rare T-cell receptor gene rearrangements are associated with poor outcome. Genes Chromosomes Cancer 48 (9): 795-805, 2009.
- Mansour MR, Abraham BJ, Anders L, et al.: Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346 (6215): 1373-7, 2014.
- Steimlé T, Dourthe ME, Alcantara M, et al.: Clinico-biological features of T-cell acute lymphoblastic leukemia with fusion proteins. Blood Cancer J 12 (1): 14, 2022.
- Weng AP, Ferrando AA, Lee W, et al.: Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306 (5694): 269-71, 2004.
- Gallo Llorente L, Luther H, Schneppenheim R, et al.: Identification of novel NOTCH1 mutations: increasing our knowledge of the NOTCH signaling pathway. Pediatr Blood Cancer 61 (5): 788-96, 2014.
- Burns MA, Place AE, Stevenson KE, et al.: Identification of prognostic factors in childhood T-cell acute lymphoblastic leukemia: Results from DFCI ALL Consortium Protocols 05-001 and 11-001. Pediatr Blood Cancer 68 (1): e28719, 2021.
- Petit A, Trinquand A, Chevret S, et al.: Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood 131 (3): 289-300, 2018.
- Trinquand A, Tanguy-Schmidt A, Ben Abdelali R, et al.: Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a Group for Research in Adult Acute Lymphoblastic Leukemia study. J Clin Oncol 31 (34): 4333-42, 2013.
- Bergeron J, Clappier E, Radford I, et al.: Prognostic and oncogenic relevance of TLX1/HOX11 expression level in T-ALLs. Blood 110 (7): 2324-30, 2007.
- van Grotel M, Meijerink JP, Beverloo HB, et al.: The outcome of molecular-cytogenetic subgroups in pediatric T-cell acute lymphoblastic leukemia: a retrospective study of patients treated according to DCOG or COALL protocols. Haematologica 91 (9): 1212-21, 2006.
- Cavé H, Suciu S, Preudhomme C, et al.: Clinical significance of HOX11L2 expression linked to t(5;14)(q35;q32), of HOX11 expression, and of SIL-TAL fusion in childhood T-cell malignancies: results of EORTC studies 58881 and 58951. Blood 103 (2): 442-50, 2004.
- Baak U, Gökbuget N, Orawa H, et al.: Thymic adult T-cell acute lymphoblastic leukemia stratified in standard- and high-risk group by aberrant HOX11L2 expression: experience of the German multicenter ALL study group. Leukemia 22 (6): 1154-60, 2008.
- Ferrando AA, Neuberg DS, Dodge RK, et al.: Prognostic importance of TLX1 (HOX11) oncogene expression in adults with T-cell acute lymphoblastic leukaemia. Lancet 363 (9408): 535-6, 2004.
- Burmeister T, Gökbuget N, Reinhardt R, et al.: NUP214-ABL1 in adult T-ALL: the GMALL study group experience. Blood 108 (10): 3556-9, 2006.
- Graux C, Stevens-Kroef M, Lafage M, et al.: Heterogeneous patterns of amplification of the NUP214-ABL1 fusion gene in T-cell acute lymphoblastic leukemia. Leukemia 23 (1): 125-33, 2009.
- Hagemeijer A, Graux C: ABL1 rearrangements in T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer 49 (4): 299-308, 2010.
- Quintás-Cardama A, Tong W, Manshouri T, et al.: Activity of tyrosine kinase inhibitors against human NUP214-ABL1-positive T cell malignancies. Leukemia 22 (6): 1117-24, 2008.
- Clarke S, O'Reilly J, Romeo G, et al.: NUP214-ABL1 positive T-cell acute lymphoblastic leukemia patient shows an initial favorable response to imatinib therapy post relapse. Leuk Res 35 (7): e131-3, 2011.
- Deenik W, Beverloo HB, van der Poel-van de Luytgaarde SC, et al.: Rapid complete cytogenetic remission after upfront dasatinib monotherapy in a patient with a NUP214-ABL1-positive T-cell acute lymphoblastic leukemia. Leukemia 23 (3): 627-9, 2009.
- Crombet O, Lastrapes K, Zieske A, et al.: Complete morphologic and molecular remission after introduction of dasatinib in the treatment of a pediatric patient with t-cell acute lymphoblastic leukemia and ABL1 amplification. Pediatr Blood Cancer 59 (2): 333-4, 2012.
- Seki M, Kimura S, Isobe T, et al.: Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat Genet 49 (8): 1274-1281, 2017.
- Nagel S, Scherr M, Kel A, et al.: Activation of TLX3 and NKX2-5 in t(5;14)(q35;q32) T-cell acute lymphoblastic leukemia by remote 3'-BCL11B enhancers and coregulation by PU.1 and HMGA1. Cancer Res 67 (4): 1461-71, 2007.
- Gutierrez A, Kentsis A, Sanda T, et al.: The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood 118 (15): 4169-73, 2011.
- Ceppi F, Gotti G, Möricke A, et al.: Near-tetraploid T-cell acute lymphoblastic leukaemia in childhood: Results of the AIEOP-BFM ALL studies. Eur J Cancer 175: 120-124, 2022.
- Zhang J, Ding L, Holmfeldt L, et al.: The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481 (7380): 157-63, 2012.
- Gutierrez A, Dahlberg SE, Neuberg DS, et al.: Absence of biallelic TCRgamma deletion predicts early treatment failure in pediatric T-cell acute lymphoblastic leukemia. J Clin Oncol 28 (24): 3816-23, 2010.
- Yang YL, Hsiao CC, Chen HY, et al.: Absence of biallelic TCRγ deletion predicts induction failure and poorer outcomes in childhood T-cell acute lymphoblastic leukemia. Pediatr Blood Cancer 58 (6): 846-51, 2012.
- Montefiori LE, Bendig S, Gu Z, et al.: Enhancer Hijacking Drives Oncogenic BCL11B Expression in Lineage-Ambiguous Stem Cell Leukemia. Cancer Discov 11 (11): 2846-2867, 2021.
- Di Giacomo D, La Starza R, Gorello P, et al.: 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T-lymphoid and myeloid immature acute leukemia. Blood 138 (9): 773-784, 2021.
- Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.
- Borowitz MJ, Béné MC, Harris NL, et al.: Acute leukaemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. International Agency for Research on Cancer, 2017, pp 179-87.
- Davies SM, Bhatia S, Ross JA, et al.: Glutathione S-transferase genotypes, genetic susceptibility, and outcome of therapy in childhood acute lymphoblastic leukemia. Blood 100 (1): 67-71, 2002.
- Krajinovic M, Costea I, Chiasson S: Polymorphism of the thymidylate synthase gene and outcome of acute lymphoblastic leukaemia. Lancet 359 (9311): 1033-4, 2002.
- Krajinovic M, Lemieux-Blanchard E, Chiasson S, et al.: Role of polymorphisms in MTHFR and MTHFD1 genes in the outcome of childhood acute lymphoblastic leukemia. Pharmacogenomics J 4 (1): 66-72, 2004.
- Schmiegelow K, Forestier E, Kristinsson J, et al.: Thiopurine methyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. Leukemia 23 (3): 557-64, 2009.
- Relling MV, Hancock ML, Boyett JM, et al.: Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 93 (9): 2817-23, 1999.
- Stanulla M, Schaeffeler E, Flohr T, et al.: Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA 293 (12): 1485-9, 2005.
- Yang JJ, Landier W, Yang W, et al.: Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol 33 (11): 1235-42, 2015.
- Relling MV, Hancock ML, Rivera GK, et al.: Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 91 (23): 2001-8, 1999.
- Moriyama T, Nishii R, Perez-Andreu V, et al.: NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet 48 (4): 367-73, 2016.
- Tanaka Y, Kato M, Hasegawa D, et al.: Susceptibility to 6-MP toxicity conferred by a NUDT15 variant in Japanese children with acute lymphoblastic leukaemia. Br J Haematol 171 (1): 109-15, 2015.
- Diouf B, Crews KR, Lew G, et al.: Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA 313 (8): 815-23, 2015.
- Yang JJ, Cheng C, Yang W, et al.: Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA 301 (4): 393-403, 2009.
- Gregers J, Christensen IJ, Dalhoff K, et al.: The association of reduced folate carrier 80G>A polymorphism to outcome in childhood acute lymphoblastic leukemia interacts with chromosome 21 copy number. Blood 115 (23): 4671-7, 2010.
- Radtke S, Zolk O, Renner B, et al.: Germline genetic variations in methotrexate candidate genes are associated with pharmacokinetics, toxicity, and outcome in childhood acute lymphoblastic leukemia. Blood 121 (26): 5145-53, 2013.
Asignación del tratamiento según el riesgo
Introducción al tratamiento según el riesgo
Los niños con leucemia linfoblástica aguda (LLA) por lo general reciben un tratamiento de acuerdo al grupo de riesgo definido a partir de características clínicas y de laboratorio. La intensidad del tratamiento necesario para curar la enfermedad varía mucho entre los subgrupos de niños con LLA. La asignación del tratamiento según el riesgo se usa en los niños con LLA a fin de evitar un tratamiento más intensivo y tóxico en los pacientes con características clínicas y biológicas favorables, que tienen probabilidad de presentar un desenlace muy bueno con un tratamiento moderado; por el contrario, se administra un tratamiento más intensivo, y posiblemente más tóxico, a los pacientes con menor probabilidad de supervivencia a largo plazo.
En algunos grupos de estudio de la LLA, como el Children´s Oncology Group (COG), se utiliza un régimen de inducción más o menos intensivo, que se determina a partir de un subconjunto de factores previos al tratamiento, mientras que en otros grupos se administran a todos los pacientes regímenes de inducción similares.
Entre los factores que el COG usa para determinar la intensidad de la inducción se incluyen los siguientes:
- Inmunofenotipo.
- Presencia o ausencia de enfermedad extramedular.
- Pretratamiento con corticoesteroides.
- Presencia o ausencia de síndrome de Down.
- Clasificación del grupo de riesgo del Instituto Nacional del Cáncer (NCI).
La clasificación del grupo de riesgo del NCI para la LLA-B estratifica el riesgo según la edad y el recuento de glóbulos blancos (GB), de la siguiente manera:
- Riesgo estándar: recuento de GB inferior a 50 000/μl y edad entre 1 y 10 años.
- Riesgo alto: recuento de GB de 50 000/μl o superior, o 10 o más años de edad.
Todos los grupos de estudio modifican la intensidad de la terapia de posinducción a partir de varios factores pronósticos, como el grupo de riesgo del NCI, el inmunofenotipo, la respuesta temprana y las alteraciones citogenéticas y genómicas. La detección de la fusión BCR::ABL1 (es decir., LLA positiva para BCR::ABL1) produce cambios inmediatos en la terapia de inducción, incluso la incorporación de un inhibidor de tirosina–cinasas, como el imatinib o el dasatinib.
La asignación del tratamiento según el riesgo requiere de la disponibilidad de factores pronósticos que predigan el desenlace de forma confiable. Para los niños con LLA se ha demostrado que varios factores tienen importancia pronóstica como los que se describen a continuación. Los factores que afectan el pronóstico se agrupan en las siguientes tres categorías:
- Características del paciente y del cuadro clínico de la enfermedad.
- Características leucémicas.
- Respuesta al tratamiento inicial.
Como en cualquier análisis de factores pronósticos, el orden relativo de significación de las variables y su interrelación a menudo depende del tratamiento, por lo tanto es necesario realizar análisis multivariantes para determinar los factores que operan de forma independiente como variables pronósticas. Debido a que los factores pronósticos dependen del tratamiento, es posible que las mejoras terapéuticas disminuyan o anulen la importancia de cualquiera de estos factores pronósticos.
Se usa un subconjunto de factores pronósticos y clínicos, expuestos más adelante, para la estratificación inicial de los niños con LLA a fin de establecer la asignación al tratamiento. Para una descripción breve de las agrupaciones pronósticas que se usan en los ensayos clínicos en curso en los Estados Unidos, consultar la sección Grupos pronósticos (riesgo) en evaluación clínica.
Para obtener información sobre factores pronósticos importantes durante la recaída, consultar la sección Factores pronósticos después de la primera recaída de la leucemia linfoblástica aguda infantil.
Factores pronósticos que afectan el tratamiento según el riesgo
Características del paciente y del cuadro clínico de la enfermedad
Las características del paciente y del cuadro clínico de la enfermedad que afectan el pronóstico son las siguientes:
- Edad en el momento del diagnóstico.
- Recuento de glóbulos blancos en el momento del diagnóstico.
- Compromiso del sistema nervioso central en el momento del diagnóstico.
- Compromiso testicular en el momento del diagnóstico.
- Síndrome de Down (trisomía 21).
- Sexo.
- Raza y etnia.
- Peso en el momento del diagnóstico y durante el tratamiento.
Edad en el momento del diagnóstico
La edad en el momento del diagnóstico tiene gran importancia pronóstica en los pacientes con LLA-B, ya que refleja las diferencias biológicas subyacentes de la LLA en diferentes grupos etarios. La edad en el momento del diagnóstico no es relevante para el pronóstico de la LLA-T.
- Lactantes (menores de 1 año).
Los lactantes con LLA tienen un riesgo especialmente alto de fracaso del tratamiento. El fracaso del tratamiento es más común en los siguientes grupos:
- Lactantes menores de 6 meses (pronóstico incluso más precario en bebés de ≤90 días).
- Lactantes con recuentos leucocitarios muy altos en el momento de la presentación inicial (>200 000–300 000 × 109/l).
- Lactantes con respuesta precaria a la profase de prednisona.
- Lactantes con un reordenamiento del gen KMT2A.
Hasta un 80 % de los lactantes con LLA exhiben una translocación de 11q23 con numerosos compañeros cromosómicos que generan un reordenamiento del gen KMT2A. El reordenamiento más frecuente es el KMT2A::AFF1 (t(4;11)(q21;q23)), pero también se observan reordenamientos de KMT2A con muchos otros compañeros de translocación. Los lactantes con leucemia y reordenamientos de KMT2A suelen tener recuentos de GB muy altos y mayor incidencia de compromiso en el SNC. Las tasas de supervivencia sin complicaciones (SSC) y supervivencia general (SG) son precarias. Las tasas de SSC y SG a 5 años son del 35 % al 40 % en los lactantes con LLA que presenta reordenamientos de KMT2A.
La frecuencia de reordenamientos del gen KMT2A es muy alta en los lactantes menores de 6 meses. Entre los 6 meses y el primer año de edad, la incidencia de reordenamientos de KMT2A disminuye, pero se mantiene significativamente más alta que la que se observa en niños de más edad. Los blastocitos de los lactantes con reordenamientos de KMT2A suelen ser negativos para CD10 y expresan índices elevados de FLT3. Por el contrario, los lactantes cuyas células leucémicas exhiben una configuración de línea germinal del gen KMT2A a menudo presentan un inmunofenotipo de células B precursoras positivas para CD10. Estos lactantes tienen un desenlace significativamente superior al de los lactantes con LLA caracterizada por reordenamientos de KMT2A.
Los lactantes negros con LLA tienen una probabilidad mucho más baja de tener reordenamientos de KMT2A que los lactantes blancos.
En una comparación del contexto de variantes somáticas en lactantes y niños de más edad que presentaban LLA con reordenamiento KMT2A se observaron diferencias significativas entre los 2 grupos. Estos resultados indican que la LLA con reordenamiento de KMT2A presenta comportamientos biológicos particulares según la edad, lo que quizás se relacione con el desenlace significativamente más precario de los lactantes.
Para obtener más información sobre los lactantes con LLA, consultar la sección Lactantes con leucemia linfoblástica aguda.
- Niños pequeños (1 a <10 años de edad)
Los niños pequeños (1 a <10 años de edad) con LLA-B tienen una tasa de supervivencia sin enfermedad (SSE) superior a la de los niños de más edad, los adolescentes y los lactantes. El mejor pronóstico de los niños pequeños se explica, al menos en parte, por la frecuencia más alta de características citogenéticas favorables en los blastocitos leucémicos, como la hiperdiploidía con 51 a 65 cromosomas, las trisomías cromosómicas favorables o la fusión ETV6::RUNX1 (t(12;21)(p13;q22), conocida en el pasado como la translocación TEL::AML1).
- Adolescentes y adultos jóvenes (≥10 años de edad)
En general, el desenlace de los pacientes de 10 años o más con LLA-B es inferior al de pacientes de 1 a 10 años. Los pacientes de entre 10 y 15 años evolucionan de manera más favorable que aquellos que se diagnostican entre los 16 y los 21 años y que se tratan con regímenes pediátricos. Sin embargo, el desenlace de los adolescentes de más edad ha mejorado de manera significativa con el tiempo. Las tasas de supervivencia a 5 años de adolescentes entre 15 y 19 años aumentaron del 36 % (1975–1984) al 78 % (2011-2017).
En varios estudios retrospectivos se ha indicado que los adolescentes de 16 a 21 años tienen un mejor desenlace cuando reciben protocolos pediátricos, en lugar de los protocolos para adultos. Para obtener más información sobre los adolescentes con LLA, consultar la sección Tratamiento de posinducción para los subgrupos específicos de leucemia linfoblástica aguda.
Recuento de glóbulos blancos en el momento del diagnóstico
Un recuento de glóbulos blancos (GB) de 50 000/µl por lo general se usa como valor de corte para definir un pronóstico favorable o desfavorable, aunque la relación entre el recuento de GB y el pronóstico es continua, no escalonada. El recuento de GB alto en el momento del diagnóstico acarrea un riesgo alto de fracaso del tratamiento en los pacientes con LLA-B, en comparación con los pacientes con recuentos de GB iniciales bajos.
La mediana de GB en el momento del diagnóstico es mucho más alta en la LLA-T (>50 000/µl) que en la LLA-B (<10 000/µl), y el efecto pronóstico del recuento de GB en el momento del diagnóstico no es uniforme en los pacientes con LLA-T.
Compromiso del sistema nervioso central en el momento del diagnóstico
La presencia o ausencia de leucemia en el sistema nervioso central (SNC) en el momento del diagnóstico tiene importancia pronóstica en los pacientes con LLA-B y LLA-T. Los pacientes sometidos a una punción lumbar diagnóstica no traumática se asignan a una de tres categorías según el número de GB/µl y la presencia o ausencia de blastocitos en una prueba con citocentrífuga de la siguiente manera.
- SNC1: líquido cefalorraquídeo (LCR) que da negativo para blastocitos en la prueba con citocentrífuga, sin importar el recuento de GB.
- SNC2: LCR con menos de 5 GB/µl y resultado positivo para blastocitos en la prueba con citocentrífuga.
- SNC3 (enfermedad en el SNC): LCR con 5 Gb/µl o más y resultado positivo para blastocitos en la prueba con citocentrífuga o signos clínicos de leucemia en el SNC (es decir, parálisis del nervio facial, compromiso del encéfalo o el ojo, o síndrome hipotalámico).
La enfermedad SNC3 en el momento del diagnóstico en los niños con LLA-B o LLA-T acarrea un riesgo más alto de fracaso del tratamiento (en el SNC y sistémico) que la enfermedad SNC1 o SNC2. La repercusión pronóstica del estado CNS2 en el momento del diagnóstico quizás difiera entre los pacientes con LLA-B y LLA-T. En algunos estudios se notificó aumento del riesgo de recaída en el SNC o una SSC inferior en los pacientes con LLA-B y estado SNC2 en el momento del diagnóstico, en comparación con los pacientes con estado SNC1, aunque otros estudios fueron contradictorios. En un análisis de 2164 pacientes con LLA-T tratados en 2 ensayos del COG consecutivos, no hubo diferencias en la SSC, la SSE ni en la incidencia acumulada de recaída entre los pacientes con estados SNC1 y SNC2.
La punción lumbar traumática (≥10 eritrocitos/µl) con blastocitos en el momento del diagnóstico se relacionó con aumento del riesgo de recaída en el SNC y un desenlace general más precario en algunos estudios, pero no en otros. Los pacientes con SNC2, SNC3 o punción lumbar traumática tienen una frecuencia más alta de características pronósticas desfavorables que aquellos con SNC1, incluso recuentos de GB significativamente más altos en el momento del diagnóstico, mayor edad en el momento del diagnóstico, un aumento de la frecuencia del fenotipo LLA-T y reordenamientos del gen KMT2A.
La mayoría de los grupos de ensayos clínicos usan un abordaje de tratamiento más intensivo para el grupo de pacientes con SNC2 y para el grupo que tiene una punción lumbar traumática; en especial se usan dosis adicionales de terapia intratecal durante la inducción.; [Nivel de evidencia B4]; [Nivel de evidencia A1]
A fin de determinar si un paciente con una punción lumbar traumática (con blastocitos) se debe tratar como un paciente del grupo SNC3, el COG utiliza un algoritmo que incluye los recuentos de GB y glóbulos rojos en el líquido raquídeo y en la sangre periférica.
Compromiso testicular en el momento del diagnóstico
El compromiso testicular evidente en el momento del diagnóstico se presenta en alrededor del 2 % de los niños, y es más frecuente en los pacientes con LLA-T que en los pacientes con LLA-B.
En los primeros ensayos de la LLA, el compromiso testicular en el momento del diagnóstico era un factor de pronóstico adverso. Sin embargo, no tiene importancia pronóstica cuando el tratamiento inicial es más intensivo. Por ejemplo, en un ensayo de la European Organization for Research and Treatment of Cancer (EORTC-58881) se notificó ausencia de importancia pronóstica adversa para el compromiso testicular evidente en el momento del diagnóstico.
No está clara la función de la radioterapia para el compromiso testicular. En un estudio del St. Jude Children's Research Hospital (SJCRH), se indica que quizás se logre un desenlace favorable con quimioterapia convencional intensiva sin radiación. El COG también adoptó esta estrategia para los niños con compromiso testicular resuelto por completo al final de la terapia de inducción. El COG incluye en el grupo de riesgo alto a los pacientes con compromiso testicular, sin importar otras características del cuadro clínico inicial, pero la mayoría de los otros grupos de ensayos clínicos grandes en los Estados Unidos y Europa no consideran la enfermedad testicular como una característica de riesgo alto.
Síndrome de Down (trisomía 21)
El desenlace de los niños con síndrome de Down y LLA suele ser inferior al de los niños sin este síndrome. Sin embargo, en algunos estudios, los pacientes con síndrome de Down evolucionaron tan bien como los que no tenían el síndrome. La SSC y la SG inferiores en los niños con síndrome de Down se relacionan con un aumento de la mortalidad relacionada con el tratamiento, y tasas más altas de fracaso de la inducción y recaída. El desenlace antileucémico inferior quizás se deba, al menos en parte, a que los pacientes con LLA y síndrome de Down tienen una frecuencia más baja de las características biológicas favorables como ETV6::RUNX1 o hiperdiploidía (51–65 cromosomas) con trisomías de los cromosomas 4 y 10.
- En un estudio retrospectivo grande de 653 pacientes con síndrome de Down y LLA, los pacientes con este síndrome tuvieron una tasa más baja de remisión completa (RC) (97 vs. 99 %, P<0,001), una incidencia acumulada más alta de recaída (26 vs. 15 %, P< 0,001) y una mortalidad relacionada con el tratamiento más alta (7 vs. <1 %, P<0,001) que los pacientes sin síndrome de Down. En los pacientes con síndrome de Down, la edad inferior a 6 años, el recuento de GB inferior a 10 000/µl y la fusión ETV6::RUNX1 (que se observó en un 8 % de los pacientes) fueron factores de predicción independientes de una SSC favorable.
- En un informe del COG de pacientes con LLA-B que carecían de reordenamientos de KMT2A, BCR::ABL1, ETV6::RUNX1 e hiperdiploidía con trisomías de los cromosomas 4 y 10, la SSC y la SG fueron similares en los niños con síndrome de Down y en aquellos sin este síndrome.
- Determinadas anomalías genómicas, como las deleciones de IKZF1, las anomalías de CRLF2 y las variantes de JAK se observan con mayor frecuencia en la LLA de los niños con síndrome de Down que en los niños sin este síndrome. Los estudios de niños con síndrome de Down y LLA indican que la presencia de deleciones de IKZF1 (pero no anomalías de CRLF2 o variantes de JAK) acarrea un pronóstico inferior.
- En un análisis retrospectivo, en el que se incluyeron 130 pacientes de LLA con reordenamiento CRLF2 y síndrome de Down, los pacientes con la firma mutacional similar a BCR::ABL1 (25% de los casos con reordenamientos de CRLF2) tuvieron un desenlace más precario, en comparación con aquellos que no presentaban la firma mutacional similar a BCR::ABL1 (tasas de SSC, 39,5 % ± 8,1 % vs. 82,0 % ± 4,4 %; cociente de riesgos instantáneos [CRI], 5,27).
- La fusión génica IGH::IGF2BP1 se presenta en alrededor del 3 % de los pacientes con síndrome de Down. Esta fusión es infrecuente en pacientes con LLA que no tienen síndrome de Down. En un análisis retrospectivo, la fusión se relacionó con un desenlace bastante favorable (tasa de SSC, 87,5 % ± 1,7 %).
Sexo
En algunos estudios, el pronóstico de las niñas con LLA es un poco mejor que el de los niños con esta enfermedad. Una de las razones del mejor pronóstico es la frecuencia de las recaídas testiculares en los niños, quienes también tienen un riesgo más alto de recaída en la médula ósea y el SNC por razones que no se comprenden bien. Mientras en algunos informes se describen los desenlaces de los niños como cercanos a los de las niñas, los ensayos clínicos más grandes y los datos del orden nacional continúan indicando tasas de supervivencia un poco inferiores para los niños.
Raza y etnia
Durante las últimas décadas, las tasas de supervivencia de los niños negros e hispanos con LLA en los Estados Unidos han sido algo menores que las de los niños blancos con esta enfermedad. En un estudio se incluyó a más de 18 031 pacientes con LLA-B y 1892 pacientes con LLA-T que tenían entre 0 y 30 años, que se trataron entre 2004 y 2019 en ensayos clínicos del COG. Las diferencias en los resultados según la raza y el origen étnico que se observaron en estudios más antiguos se mantuvieron con las terapias más contemporáneas. Se observaron diferencias en los resultados según la raza y el origen étnico en los pacientes con LLA-B, pero no en los pacientes con LLA-T. En el estudio también se observó una disparidad más amplia en la SG versus la SSC en los pacientes con LLA-B, lo que indica que las diferencias podrían ser mayores en el entorno de una enfermedad recidivante versus una enfermedad recién diagnosticada. El análisis multivariante que incorporó el ajuste para los factores pronósticos de la enfermedad (por ejemplo, edad y recuento de glóbulos blancos, grupo de riesgo citogenético, estado del SNC) y la situación respecto al seguro médico atenuó en gran medida el aumento del riesgo de una SSC inferior en los pacientes hispanos. Sin embargo, los mismos ajustes no atenuaron la SSC inferior en los niños negros no hispanos.
Los factores relacionados con la raza y la etnia que afectan la supervivencia son los siguientes:
- Subtipo de LLA. La razón de pronóstico más favorable en los niños blancos y asiáticos que en los niños negros e hispanos se explica, al menos de manera parcial, por la variedad de subtipos de LLA. Por ejemplo, los niños negros tienen una mayor incidencia relativa de LLA-T, menores tasas de los subtipos genéticos favorables de LLA-B y tasas más altas de la translocación TFC3::PBX1 (t(1;19)). Los niños hispanos y latinos tienen una frecuencia más alta de reordenamientos de CRLF2 y deleciones de IKZF1.
- Cumplimiento terapéutico. Es posible que las diferencias en el desenlace también obedezcan al cumplimiento terapéutico, como se ilustra en un estudio de cumplimiento con el mantenimiento de mercaptopurina (6-MP) oral. En el primer informe del estudio, el riesgo de recaída aumentó según el grado de cumplimiento de los niños hispanos en comparación con los niños blancos no hispanos, incluso cuando se ajustó por otras variables conocidas. Sin embargo, aún con tasas de cumplimiento del 90 % o más, las tasas de recaída siguieron siendo elevadas en los niños hispanos. En el segundo informe del estudio, las tasas de cumplimiento fueron significativamente más bajas en los pacientes estadounidenses de origen asiático y afroamericanos que en los pacientes blancos no hispanos. Un porcentaje más alto de pacientes de estos grupos étnicos exhibieron tasas de cumplimiento inferiores al 90 %, lo que se relacionó con un aumento del riesgo de recaída de 3,9 veces.
- Variaciones genómicas relacionadas con la ascendencia. Las variaciones genómicas relacionadas con la ascendencia también contribuyen a las desigualdades raciales y étnicas en la incidencia y el desenlace de los pacientes con LLA. Por ejemplo, la presencia diferencial de polimorfismos específicos en los diferentes grupos raciales o étnicos contribuye a las desigualdades en los desenlaces, como se ilustra por la frecuencia de polimorfismos de un solo nucleótido en el gen ARID5B, que es más frecuente en hispanos y se relaciona con susceptibilidad a la LLA y riesgo de recaída. En un estudio de asociación de genoma completo, la variante GATA3, rs3824662, se relacionó con un incremento del riesgo de presentar LLA similar a BCR::ABL1 (similar a Ph). Los pacientes con esta variante presentaban un incremento del riesgo de ERM al final de la inducción y un mayor riesgo de recaída. El alelo de riesgo rs3824662 se vincula con la ascendencia genética de las personas indígenas de las Américas. La frecuencia del alelo de riesgo fue del 52 % en los pacientes guatemaltecos, del 40 % en los pacientes estadounidenses de origen hispano y del 14 % en los pacientes con ascendencia europea.
Peso en el momento del diagnóstico y durante el tratamiento
En estudios sobre el efecto de la obesidad en el desenlace de la LLA se obtuvieron resultados variados. En la mayoría de estos estudios, la obesidad se define como un peso superior al percentil 95 para la edad y la talla.
- En tres estudios no se demostró ningún efecto independiente de la obesidad en la SSC.[Nivel de evidencia B4]; [Nivel de evidencia C2]
- En 2 estudios se observó que la obesidad es un factor pronóstico independiente solo en pacientes mayores de 10 años o en quienes tienen una enfermedad de riesgo intermedio o riesgo alto.[Nivel de evidencia C2]
- El COG informó sobre el efecto de la obesidad en el desenlace de 2008 niños (14 % obesos) que participaron en un ensayo de LLA de riesgo alto (CCG-1961 [NCT00002812]).[Nivel de evidencia B4] Se encontró que la obesidad fue una variable independiente para un desenlace inferior, en comparación con los pacientes sin obesidad (tasas de SSC a 5 años, 64 vs. 74 %; P = 0,002). Sin embargo, los pacientes obesos en el momento del diagnóstico, que perdieron peso durante el tratamiento previo al mantenimiento, tuvieron desenlaces similares a los de los pacientes con peso normal en el momento del diagnóstico.
- En un estudio retrospectivo de pacientes tratados en una sola institución, la obesidad en el momento del diagnóstico se relacionó con un aumento del riesgo de presentar ERM al final de la inducción y una SSC inferior.[Nivel de evidencia C2]
- En otro estudio retrospectivo de 373 pacientes tratados en una sola institución, el índice de masa corporal (IMC) en el momento del diagnóstico no se relacionó con una ERM en los días 19 ni 46, tampoco con una incidencia acumulada de recaída ni con la SSC. La SG fue inferior en los pacientes con un IMC alto, principalmente por la mortalidad relacionada con el tratamiento y el rescate precario después de la recaída.[Nivel de evidencia C1]
- En un estudio, la obesidad en el momento del diagnóstico se relacionó con un incremento de la toxicidad y con la administración incompleta de asparaginasa, en particular en niños de más edad y adolescentes.
- En un estudio de 388 pacientes de 15 a 50 años tratados con los regímenes del consorcio de LLA del Dana-Farber Cancer Institute (DFCI), un IMC más elevado se relacionó con tasas más altas de recaída y mortalidad no relacionada con la recaída, así como una SG más corta. Un IMC más elevado se asoció con tasas más altas de hepatotoxicidad e hiperglucemia. El efecto deletéreo del IMC elevado fue más pronunciado en los pacientes de más edad. Entre los pacientes de 15 a 29 años en el momento del diagnóstico (n = 254), la tasa de SG a 4 años fue del 73 % para aquellos con un IMC elevado en comparación con el 83 % para aquellos con un IMC en el intervalo de referencia (P = 0,09).
En un estudio de 762 pacientes pediátricos con LLA (edad, 2–17 años), el Dutch Childhood Oncology Group concluyó que el peso insuficiente en el momento del diagnóstico (definido como un puntaje de desviación estándar del IMC< -1,8; 8 % de la población) acarreó casi el doble de riesgo de recaída, en comparación con el peso suficiente (después del ajuste por grupo de riesgo y edad), aunque esto no se tradujo en diferencias en la SSC o la SG. Los pacientes que disminuyeron su IMC durante las primeras 32 semanas de tratamiento presentaron tasas de recaída parecidas a otros pacientes, pero su SG fue significativamente peor, sobre todo debido a unas tasas de rescate más precarias después de la recaída.
Características leucémicas
Las características de las células leucémicas que afectan el pronóstico son las siguientes:
- Inmunofenotipo.
- Alteraciones citogenéticas o genómicas.
Inmunofenotipo
En la revisión de 2016 de la clasificación de las neoplasias mieloides y leucemias agudas de la Organización Mundial de la Salud (OMS), se clasifica la LLA como leucemia linfoblástica B o leucemia linfoblástica T, con subdivisiones según las características moleculares. Para obtener más información, consultar la sección Diagnóstico.
Es posible que las leucemias linfoblásticas B o T coexpresen antígenos mieloides. Es necesario diferenciar estos casos de la leucemia de linaje ambiguo.
- Leucemia linfoblástica aguda de células B (leucemia linfoblástica B según la OMS)
Antes de 2008, la OMS clasificaba la leucemia linfoblástica B como leucemia linfoblástica de células B precursoras y este término todavía se usa con frecuencia en la bibliografía de LLA infantil para diferenciarla de la LLA de células B maduras. La LLA de células B maduras ahora se denomina leucemia de Burkitt y exige un tratamiento diferente del que se administra para la LLA-B (LLA de células B precursoras).
La LLA-B, definida por la expresión de CD19, HLA-DR, CD79a citoplasmático, y otros antígenos de las células B; representa entre el 80 % y el 85 % de los casos de LLA infantil. Alrededor del 90 % de los casos de LLA-B expresan el antígeno de superficie CD10 (antes conocido como antígeno común de la LLA). La ausencia de CD10 a menudo se relaciona con reordenamientos de KMT2A, en especial, t(4;11)(q21;q23), y un desenlace precario. No está claro si la negatividad para CD10 tiene una importancia pronóstica independiente cuando no hay un reordenamiento del gen KMT2A.
Los principales subtipos inmunofenotípicos de la LLA-B son los siguientes:
- Leucemia linfoblástica aguda de células B comunes (positiva para CD10, sin inmunoglobulina [Ig] de superficie ni citoplasmática)
Cerca de tres cuartos de los pacientes con LLA-B exhiben el inmunofenotipo de células B precursoras comunes y tienen el mejor pronóstico. Los pacientes con características citogenéticas favorables casi siempre exhiben un inmunofenotipo de células B precursoras comunes.
- Leucemia linfoblástica aguda pro-B (negativa para CD10, sin Ig de superficie ni citoplasmática)
Cerca del 5 % de los pacientes presenta el inmunofenotipo pro-B. El inmunofenotipo pro-B es el más común en los lactantes y a menudo se relaciona con reordenamientos del gen KMT2A.
- Leucemia linfoblástica aguda pre-B (presencia de Ig citoplasmática)
Las células leucémicas de los pacientes con LLA pre-B contienen Ig citoplasmática y un 25 % exhiben la translocación t(1;19)(q21;p13) y la fusión TCF3::PBX1.
Cerca del 3 % de los pacientes tiene una LLA pre-B transicional con expresión en la superficie de Ig de cadena pesada sin expresión de Ig de cadena ligera, además de compromiso del gen MYC y tipo morfológico L3. Los pacientes que exhiben este fenotipo responden bien al tratamiento que se usa para la LLA-B.
- Leucemia linfoblástica aguda de células B maduras (linfoma o leucemia de Burkitt).
Cerca del 2 % de los pacientes presentan al inicio una leucemia de células B maduras (expresión de Ig de superficie, por lo general, de tipo morfológico L3 según los criterios French-American-British y una translocación 8q24 que compromete el gen MYC), llamada también leucemia de Burkitt. El tratamiento de la LLA de células B maduras se basa en el abordaje del linfoma no Hodgkin y es completamente diferente al tratamiento de la LLA-B. Los casos poco frecuentes de leucemia de células B maduras que carecen de Ig de superficie, pero exhiben un tipo morfológico de L3 con translocaciones del gen MYC también se deben tratar como una leucemia de células B maduras. Para obtener más información acerca del tratamiento de niños con linfoma o leucemia de células B maduras y linfoma o leucemia de Burkitt, consultar Tratamiento del linfoma no Hodgkin infantil.
Se ha notificado un pequeño número de casos de leucemias con translocación IG::MYC que exhiben un inmunofenotipo de células B precursoras (por ejemplo, ausencia de expresión de CD20 y expresión de Ig de superficie). Estos casos se presentaron en niños y adultos. Al igual que el linfoma o la leucemia de Burkitt, este tipo predominó en los varones y la mayoría de los pacientes exhibía un tipo morfológico L3. Estos casos carecían de variantes de genes alterados de manera recurrente en el linfoma de Burkitt (por ejemplo, ID3, CCND3 o MYC), aunque eran comunes las variantes de los genes RAS (frecuentemente alterados en la LLA-B). La importancia clínica de las leucemias con translocaciones IG::MYC de fenotipo y características moleculares de células B precursoras exige más estudio.
- Leucemia linfoblástica aguda de células B comunes (positiva para CD10, sin inmunoglobulina [Ig] de superficie ni citoplasmática)
- Leucemia linfoblástica aguda de células T.
La leucemia linfoblástica aguda de células T (LLA-T) se define por la expresión de antígenos de las células T (CD3 citoplasmático, CD7 y CD2 o CD5) en los blastocitos leucémicos. A menudo, la LLA-T se relaciona con un conjunto de características clínicas, como las siguientes:
- Sexo masculino.
- Edad más avanzada.
- Leucocitosis.
- Masa mediastínica.
En la actualidad, los niños con LLA-T tienen un desenlace similar al de los niños con LLA-B cuando se usan abordajes apropiados de tratamiento intensivo, pero en el pasado esto no era lo habitual.
Se conocen pocos factores pronósticos aceptados en general para los pacientes con LLA-T. Los datos sobre la importancia pronóstica del recuento leucocitario en el momento de la presentación inicial de la LLA-T son contradictorios. La presencia o ausencia de una masa mediastínica en el momento del diagnóstico no tiene importancia pronóstica. En los pacientes que tienen una masa mediastínica, la tasa de regresión de la masa carece de importancia pronóstica.
Leucemia linfoblástica aguda de células T precursoras tempranas
La leucemia linfoblástica aguda (LLA) de células T precursoras tempranas (TPT), un subconjunto particular de la LLA-T infantil, se definió inicialmente a partir de la identificación de casos de LLA-T con perfiles de expresión génica muy parecidos a los perfiles de expresión de células T precursoras tempranas normales. El subconjunto de casos de LLA-T identificado mediante estos análisis representó el 13 % de todos los casos, y se caracterizaron por un inmunofenotipo distintivo (negatividad para CD1a y CD8, expresión débil de CD5 y coexpresión de marcadores de células madre o mieloides).
En los informes preliminares de la LLA TPT se indicó que este subconjunto de pacientes tiene un pronóstico más precario que el de los pacientes con LLA-T. Además, en algunos estudios se notificó que estos pacientes presentan una respuesta temprana más lenta y una frecuencia más alta de fracaso de la inducción. En otros estudios se han observado desenlaces más favorables en los pacientes con LLA TPT, como en el estudio del U.K. Medical Research Council en el que se demostró que los subgrupos de pacientes con LLA TPT no presentaban tasas de SSC a 5 años significativamente inferiores a las de los pacientes con LLA sin TPT (76 vs. 84 %). Así mismo, en el ensayo del COG AALL0434 [NCT00408005] , el estado de TPT no tuvo un efecto estadísticamente significativo sobre la SSE (cociente de riesgos instantáneos, 0,99; IC 95 %, 0,59–1,67; P = 0,981) en los análisis multivariantes. Se necesitan más estudios en otras cohortes de pacientes para establecer de manera definitiva la importancia pronóstica de la LLA de células T precursoras tempranas, pero la mayoría de los grupos de tratamiento de LLA no cambian el abordaje terapéutico a partir del estado de células T precursoras tempranas.
- Expresión de antígenos mieloides.
Hasta un tercio de los pacientes con LLA infantil tienen células leucémicas que expresan antígenos de superficie mieloide. La expresión de un antígeno mieloide se relaciona con subgrupos específicos de LLA, en especial, tipos que exhiben reordenamientos de KMT2A,ETV6::RUNX1, y BCR::ABL1. Los pacientes con LLA-B con reordenamientos que afectan el gen ZNF384 a menudo expresan un antígeno mieloide. La expresión de un antígeno de superficie mieloide no tiene importancia pronóstica adversa independiente.
Para obtener información sobre la leucemia de linaje ambiguo, consultar la sección Clasificación de la OMS de 2016 para las leucemias agudas de linaje ambiguo.
Alteraciones citogenéticas o genómicas
Para obtener información sobre las características citogenéticas y genómicas de la LLA-B y la LLA-T, así como los polimorfismos génicos en las vías metabólicas de los fármacos, consultar la sección Características citogenéticas y genómicas de la leucemia linfoblástica aguda infantil.
Respuesta al tratamiento inicial
La rapidez de la destrucción de las células leucémicas después del inicio del tratamiento y el grado de enfermedad residual al final de la inducción se relacionan con el resultado a largo plazo. Esto se explica porque la sensibilidad farmacológica de las células leucémicas y el comportamiento farmacodinámico y farmacogenómico del huésped afectan la respuesta terapéutica, y la respuesta temprana tiene gran importancia pronóstica. Las siguientes son algunas de las formas que se han usado para evaluar la respuesta al tratamiento de la células leucémicas:
- Evaluación de la enfermedad residual mínima en la médula ósea al final de la inducción y al final de la consolidación.
- Respuestas en la médula ósea en el día 7 y el día 14.
- Respuesta en la sangre periférica a la profase de corticoesteroides.
- Respuesta en la sangre periférica a la terapia de inducción multifarmacológica.
- Enfermedad residual mínima en la sangre periférica antes del final de la inducción (días 8 y 15).
- Leucemia persistente al final de la inducción (fracaso de la inducción).
Evaluación de la enfermedad residual mínima en la médula ósea al final de la inducción y al final de la consolidación
La evaluación morfológica de la leucemia residual en la sangre o la médula ósea a menudo es complicada y relativamente insensible. Tradicionalmente, se ha usado un límite del 5 % de los blastocitos en la médula ósea (detectados por microscopía óptica) para determinar el estado de la remisión. Esto corresponde a un índice de 1 en 20 células malignas. Para detectar concentraciones más bajas de células leucémicas en sangre o médula ósea, se necesitan técnicas especializadas. Estas incluyen pruebas de reacción en cadena de la polimerasa (PCR), que identifican reordenamientos génicos únicos de la Ig o el receptor de las células T y transcritos de fusión producidos por las translocaciones cromosómicas; o ensayos de citometría de flujo, que detectan inmunofenotipos específicos de la leucemia. Dichas técnicas permiten hallar 1 célula leucémica entre 100 000 células normales, y detectan habitualmente una enfermedad residual mínima (ERM) de 1 en 10 000 células (1 × 10-4 o 0,01 %). Las técnicas nuevas de secuenciación de alto rendimiento (HTS) de los reordenamientos génicos de Ig o del receptor de células T aumentan la sensibilidad para la detección de la ERM de 1 en 1 millón de células (1 × 10-6 o 0,0001 %).
En múltiples estudios se demostró que la ERM al final de la inducción es un factor pronóstico independiente de importancia para el desenlace en niños y adolescentes con LLA de linaje B. La respuesta según la ERM permite diferenciar el desenlace para subconjuntos de pacientes definidos por la edad, el recuento leucocitario y las anomalías citogenéticas. En general, los pacientes con los índices más altos de ERM al final de la inducción tienen un pronóstico más precario que aquellos con índices más bajos o indetectables. Sin embargo, el riesgo absoluto de recaída de acuerdo con el índice de ERM específico varía según el subtipo genético. Por ejemplo, con cualquier índice detectable de ERM al final de la inducción, los pacientes con características citogenéticas favorables, como ETV6::RUNX1 o hiperdiploidía alta, tienen un riesgo absoluto de recaída posterior más bajo que otros pacientes, mientras que los pacientes con características citogenéticas de riesgo alto tienen un riesgo absoluto de recaída posterior más alto en comparación con otros pacientes. Esta observación quizás tenga repercusiones importantes al formular esquemas de clasificación del riesgo a partir de la ERM.
Casi todos los grupos usan la ERM al final de la inducción como un factor que determina la intensidad del tratamiento de posinducción. A los pacientes con índices más altos de ERM (por lo general del 0,1 % al 0,01 %) se les administran tratamientos más intensivos.; [Nivel de evidencia B4]
En un estudio de 619 niños con LLA, se comparó la utilidad pronóstica de la ERM evaluada mediante citometría de flujo y mediante el ensayo de HTS más sensible. Con el valor de corte de 0,01 % para la ERM al final de la inducción, la HTS permitió identificar casi un 30 % más de casos como positivos (es decir, >0,01 %). Los pacientes que obtuvieron un resultado positivo en la HTS, pero un resultado negativo en la citometría de flujo, exhibieron un pronóstico intermedio en comparación con los pacientes con resultados congruentes en los dos métodos, ya sea ambos positivos o negativos. Los pacientes que cumplieron con los criterios de LLA de riesgo estándar con una ERM indetectable mediante la HTS exhibieron un pronóstico especialmente favorable (SSC a 5 años, 98,1 %).
Los índices de ERM obtenidos 10 a 12 semanas después de comenzar el tratamiento (final de la consolidación) también demostraron ser de importancia pronóstica. Los pacientes con valores altos de ERM al final de la consolidación tuvieron una SSC significativamente menor en comparación con otros pacientes.
- Leucemia linfoblástica aguda de células B. A partir de la evaluación de la ERM en dos momentos (al final de la inducción y al final de la consolidación), los pacientes con LLA-B se clasifican en los siguientes tres subgrupos de pronóstico:
- ERM baja o indetectable al final de la inducción: mejor pronóstico.
- ERM alta al final de la inducción, pero ERM baja o negativa al final de la consolidación: pronóstico intermedio
- ERM alta al final de la consolidación (semana de tratamiento 10–12): peor pronóstico. El efecto de la ERM al final de la consolidación sobre el pronóstico está modulado por los criterios de riesgo del NCI: los pacientes de riesgo alto del NCI con ERM alta al final de la consolidación tienen tasas de SSE más bajas que los pacientes de riesgo estándar del NCI con índices similares de ERM al final de la consolidación.
- Leucemia linfoblástica aguda de células T. Hay menos estudios sobre la importancia pronóstica de la ERM en los pacientes con LLA-T. El grupo United Kingdom Acute Lymphoblastic Leukaemia (UKALL) notificó que los pacientes de LLA-T con ERM indetectable al final de la inducción exhibieron desenlaces excelentes, mientras que los pacientes con índices muy altos de ERM (>5 %) al final de la inducción exhibieron un pronóstico precario. Sin embargo, no se encontró ninguna relación entre el índice de ERM al final de la inducción y el riesgo de recaída para el resto de los pacientes con LLA-T. El consorcio de LLA del DFCI también notificó que los pacientes de LLA-T con una ERM muy baja al final de la inducción (<10-4) tuvieron un desenlace muy favorable.
En otro estudio también se indicó que la ERM en un momento posterior quizás acarree mayor importancia pronóstica en la LLA-T. En el ensayo de la Associazione Italiana di Ematologia e Oncologia Pediatrica (AIEOP) ALL-BFM-2000 (NCT00430118), el estado de la ERM en el día 78 (semana 12) fue el predictor de recaída más importante en los pacientes con LLA-T. Los pacientes con ERM detectable al final de la inducción que tenían un resultado negativo para la ERM en el día 78, por lo general exhibieron un pronóstico favorable, de manera similar a los pacientes con un resultado negativo para la ERM en un momento más temprano, al final de la inducción. En el estudio COG AALL1231 se confirmó la importancia pronóstica de la ERM en la médula ósea al final de la consolidación en los pacientes con LLA-T.
Las mediciones de la ERM, junto con otras características del cuadro clínico inicial, también se han usado para identificar subconjuntos de pacientes con un riesgo de recaída muy bajo. El COG notificó un pronóstico muy favorable (SSC a 5 años del 97 % ± 1 %) para los pacientes con un fenotipo de células B precursoras, edad y recuento leucocitario de riesgo estándar del NCI, estado SNC1 y anomalías citogenéticas favorables (hiperdiploidía alta con trisomías favorables o la fusión ETV6::RUNX1) que tenían índices de ERM inferiores al 0,01 % en el día 8 (en sangre periférica) y al final de la inducción (en médula ósea). Los desenlaces excelentes en los pacientes con ERM baja al final de la inducción persisten más de 10 años después del diagnóstico.
Se observó que la modificación del tratamiento a partir de la determinación de la ERM mejora el desenlace.
- En el estudio UKALL2003 (NCT00222612) se demostró que la reducción del tratamiento (es decir, 1 en lugar de 2 cursos de intensificación diferida) no afectó de manera adversa el desenlace de los pacientes que no eran de riesgo alto con una ERM favorable al final de la inducción.[Nivel de evidencia B1] En un ensayo controlado aleatorizado (UKALL2003) también se demostró mejora de la SSC para los pacientes de riesgo estándar y de riesgo intermedio que recibieron tratamiento intensificado después de una ERM al final de la inducción superior al 0,01 % (tasas de SSC a 5 años, 89,6 % para tratamiento intensificado vs. 82,8 % para tratamiento estándar).
- En el ensayo neerlandés AAL10 se estratificó a los pacientes en tres grupos de riesgo de acuerdo con la ERM después del primer mes de tratamiento y del segundo ciclo de quimioterapia, de la siguiente manera:[Nivel de evidencia B4]
- Riesgo estándar (ERM baja después del primer mes de tratamiento).
- Riesgo moderado (ERM alta después del primer mes de tratamiento, ERM baja después del segundo ciclo de quimioterapia).
- Riesgo alto (ERM alta después del segundo ciclo de quimioterapia).
En comparación con los ensayos previos dirigidos por el mismo grupo, el tratamiento fue menos intensivo para el grupo de pacientes de riesgo estándar, y más intensivo para los pacientes de riesgo moderado y riesgo alto. La tasa de SSC a 5 años (87 %) y la tasa de SG (92 %) fueron superiores a las de los estudios neerlandeses anteriores.
- En el ensayo 05-001 del DFCI ALL Consortium, los pacientes de LLA-B con ERM alta al final de la inducción (definida como ≥1 × 10-3) se clasificaron como de riesgo muy alto, con independencia de que presentaran otras características. Estos pacientes recibieron quimioterapia citotóxica intensificada en la columna vertebral. La SSE a 5 años de estos pacientes fue del 77 %, significativamente superior al desenlace de dichos pacientes en ensayos anteriores, donde no se usó la ERM para estratificar el tratamiento.
Respuestas en la médula ósea el día 7 y el día 14
Los pacientes que exhiben una reducción rápida de las células leucémicas hasta menos del 5 % en la médula ósea al cabo de 7 o 14 días del inicio de la quimioterapia multifarmacológica exhiben un pronóstico más favorable que los pacientes con una eliminación más lenta de las células leucémicas de la médula ósea. En general, las evaluaciones de la ERM al final de la terapia de inducción han reemplazado las evaluaciones morfológicas de los días 7 y 14 como indicadores pronósticos de la respuesta al tratamiento, porque estos últimos pierden su importancia pronóstica en los análisis multivariantes una vez que se incluye la ERM.
Respuesta en la sangre periférica a la profase de corticoesteroides
Los pacientes con una reducción del recuento de blastocitos periféricos hasta menos de 1000/µl después de una profase de inducción de 7 días con prednisona y una dosis de metotrexato intratecal (buena respuesta a la prednisona) exhiben un pronóstico más favorable que los pacientes con recuentos de blastocitos periféricos que superan 1000/µl (respuesta precaria a la prednisona). La respuesta precaria a la prednisona se observa en menos del 10 % de los pacientes. La estratificación del tratamiento en los protocolos de los ensayos clínicos del grupo Berlin-Frankfurt-Münster (BFM) tradicionalmente se basaba, en parte, en la respuesta temprana a la profase de prednisona de 7 días (administrada justo antes del inicio de la inducción multifarmacológica a la remisión). En el ensayo en curso que lleva a cabo ese grupo todavía se usa la respuesta a la prednisona para estratificar a los pacientes de LLA-T según el riesgo, pero no a los pacientes con LLA-B.
Respuesta en la sangre periférica a la terapia de inducción multifarmacológica
Los pacientes con células leucémicas circulantes persistentes después de 7 a 10 días de iniciar la quimioterapia multifarmacológica tienen un aumento del riesgo de recaída, en comparación con aquellos que ya no tienen blastocitos periféricos al cabo de 1 semana de tratamiento. Se encontró que la tasa de eliminación de los blastocitos periféricos tiene importancia pronóstica en la LLA de células T y de linaje B.
Enfermedad residual mínima en la sangre periférica antes del final de la inducción (días 8 y 15)
También se evaluó la ERM en sangre periférica una semana después del inicio de la quimioterapia multifarmacológica de inducción como un factor pronóstico de la respuesta temprana al tratamiento.
- En un estudio del COG en el que participaron casi 2000 niños con LLA, la presencia de ERM en la sangre periférica en el día 8 se relacionó con un pronóstico adverso. Los índices de ERM elevados se relacionaron con un desenlace progresivamente más precario.
- En el análisis multivariante, la ERM al final de la terapia de inducción fue el factor pronóstico más potente, pero la ERM en la sangre periférica el día 8 mantuvo su importancia pronóstica, así como el grupo de riesgo del NCI y la presencia de trisomías favorables. En un estudio más pequeño se evaluó la importancia pronóstica de la ERM en la sangre periférica en el día 15 después de una semana de una profase de corticoesteroides y una semana de terapia de inducción multifarmacológica. En este estudio también se observó la importancia multivariante de los índices de ERM en la sangre periférica al cabo de una semana de terapia de inducción multifarmacológica.
En ambos estudios se identificó a un grupo de pacientes que alcanzó índices bajos de ERM después de una semana de terapia de inducción multifarmacológica y que después presentó una tasa baja de fracaso del tratamiento.
Leucemia persistente al final de la inducción (fracaso de la inducción)
Casi todos los niños con LLA alcanza una remisión morfológica completa al final del primer mes de tratamiento. En el 1 % al 2 % de los niños con LLA se observa la presencia de más de un 5 % de linfoblastos en la evaluación morfológica al final de la fase de inducción.
Las características relacionadas con un riesgo más alto de fracaso de la inducción son las siguientes:
- Fenotipo de células T.
- Recuentos de GB más altos en el momento del diagnóstico para los pacientes con LLA-B.
- Edad avanzada.
- Características biológicas desfavorables.
- Reordenamiento de KMT2A.
- Reordenamiento BCR::ABL1 (antes del uso de inhibidores de tirosina–cinasas).
- Reordenamiento de PDGFRB (con mayor frecuencia EBF1::PDGFRB), que a menudo se vincula con el subtipo similar a BCR::ABL1. Estos pacientes representan menos del 1 % de los casos de LLA-B en niños, pero suponen hasta el 10 % de los casos de fracaso de la inducción. En un estudio retrospectivo, 43 de 49 pacientes (88 %) con fusiones de PDGFRB presentaron un índice de ERM al final de la inducción de más del 1 %.
En un estudio retrospectivo grande, la tasa de SG de los pacientes con fracaso de la inducción fue de solo el 32 %. Sin embargo, la heterogeneidad clínica y biológica fue significativa. Se observó un desenlace relativamente favorable en los pacientes con LLA-B de entre 1 y 5 años de edad sin características citogenéticas adversas (reordenamiento de KMT2A o BCR::ABL1). Este grupo presentó una tasa de supervivencia a 10 años que superó el 50 % y, en este subconjunto el trasplante de células madre hematopoyéticas (TCMH) durante la primera remisión no se relacionó con ninguna ventaja de supervivencia, en comparación con la quimioterapia sola. Los pacientes con los resultados más precarios (supervivencia a 10 años <20 %) fueron aquellos de 14 a 18 años de edad, o los que tenían la fusión BCR::ABL1 o reordenamiento de KMT2A. Los pacientes con LLA-B menores de 6 años y los pacientes con LLA-T (con independencia de la edad) exhiben mejores desenlaces si reciben un TCMH alogénico después de lograr una RC, en comparación con los pacientes que reciben tratamiento adicional con quimioterapia sola. Sin embargo, en el estudio del COG AALL0434 (NCT00408005) , no se observó ninguna ventaja del TCMH durante la primera RC de pacientes de LLA-T con fracaso de la inducción (definido como médula M3 al final de la inducción). En este estudio, los pacientes de LLA-T se asignaron para recibir nelarabina durante varias fases de tratamiento posinducción y dosis altas de metotrexato durante la primera fase de mantenimiento provisional. La tasa de SSC a 5 años de estos pacientes fue del 53,1 %, sin diferencias significativas entre los que se sometieron a TCMH durante la primera RC (n = 20) y los que no se sometieron (n = 23) (P = 0,42).
Citometría de flujo versus evaluación morfológica
Ahora se está integrando la ERM con la evaluación morfológica para determinar la respuesta a la terapia de inducción a partir de estudios en los que se observó que los pacientes con índices de ERM superiores al 5 %, a pesar de presentar una RC morfológica, tuvieron resultados similares a los de los pacientes con fracaso de la inducción desde el punto de vista morfológico.
- En el estudio UKALL2003 (NCT00222612), 59 de 3113 pacientes (1,9 %) presentaron fracaso de la inducción en la evaluación morfológica.
- La tasa de SSC a 5 años fue del 51 %, y la tasa de SG fue del 58 %.
- El 2,3 % de los pacientes presentó una remisión morfológica, pero tenían una ERM ≥5 % medida con PCR cuantitativa en tiempo real del receptor de células T-IgH. Este grupo tuvo una tasa de SSC del 47 %, similar a la de aquellos con fracaso de la inducción en la evaluación morfológica.
- Los autores indicaron que el uso de los criterios morfológicos y de la ERM para definir el fracaso de la inducción permitiría identificar con mayor precisión a los pacientes con desenlaces precarios.
- En un estudio de 9350 pacientes inscritos en ensayos clínicos del COG entre 2004 y 2014 se compararon las características de los pacientes y sus desenlaces clasificados por características morfológicas (M1 vs. M2/M3) y estado de la ERM evaluada por citometría de flujo (<5 vs. ≥5 %). La remisión morfológica (estado M1) se logró en el 98,6 % de los pacientes con LLA-B y en el 93,8 % de los pacientes con LLA-T al final de la terapia de inducción.
- La evaluación morfológica y la ERM coincidieron en el 97,4 % de los niños. Sin embargo, solo el 87,3 % de los pacientes con LLA-T se clasificaron como M1 y con ERM <5 %, mientras que el 97,8 % de los pacientes con LLA-B obtuvieron una remisión coincidente.
- Alrededor del 20 % los pacientes (40 de 202) con morfología M2/M3 tuvieron una ERM <5 %. Los pacientes de LLA-B con morfología M2/M3, pero ERM <5 %, tuvieron una tasa de SG a 5 años del 72,7 %, que fue inferior a la de los pacientes que coincidieron en la remisión (tasa de SG a 5 años, 93,8 %), pero superior a la de los pacientes con médula de tipo M3 (tasa de SG a 5 años, 43,4 %).
- Entre los pacientes de LLA-B y LLA-T con médula de tipo M1, el 0,9 % de los pacientes con LLA-B y el 6,9 % de los pacientes con LLA-T tuvieron una ERM ≥5 %. Su desenlace se comparó al de los pacientes con médula de tipo M1 y ERM <5 %, y se muestra en el Cuadro 5 a continuación.
- En el Cuadro 5 se muestra que en los niños de LLA-B con médula de tipo M1 y ERM ≥5 %, la tasa de SSC a 5 años fue muy inferior a la de los niños con remisión coincidente (59,1 vs. 87,1 %), pero fue superior a la de niños que no estaban en remisión coincidente (M2 con ERM ≥5 %: tasa de SG a 5 años, 39,1 %).
- La repercusión en la SSC de una ERM ≥5 % en niños con LLA-B en remisión morfológica estuvo determinada por los pacientes de riesgo alto del NCI, dado que no hubo ninguna diferencia marcada en la SSC entre los pacientes de riesgo estándar del NCI en remisión morfológica con ERM ≥5 % o sin esta.
- También se observaron tasas de SSC inferiores en pacientes de LLA-T con médula M1 y ERM ≥5 %, en comparación con aquellos en remisión coincidente (87,6 vs. 80,3 %). Sin embargo, el desenlace de los pacientes de LLA-T que no estaban en remisión (ya fuera morfológica, de ERM o ambas) fue superior a la de los pacientes de LLA-B comparables.
- Los factores predictivos de ERM discordante (≥5 %) para pacientes en remisión morfológica al final de la inducción incluyeron la edad de 10 años o más, el recuento de GB de 50 000/µl o superior en el momento de la presentación inicial, las características citogenéticas neutras o desfavorables y la LLA TPT (para pacientes con LLA-T).
| Desenlace | M1/ERM <5 % | Valor de Pb | M1/ERM ≥5 % | Valor de Pc | M2/ERM ≥5 % | |
|---|---|---|---|---|---|---|
| RA = riesgo alto; ERM = enfermedad residual mínima; RE = riesgo estándar. | ||||||
| aAdaptación de Gupta et al. | ||||||
| bValor de P para la comparación de M1/ERM <5 % con M1/ERM ≥5 %. | ||||||
| cValor de P para la comparación de M1/ERM ≥5 % con M1/ERM ≥ 5 %. | ||||||
| Tasas de supervivencia sin complicaciones: | ||||||
| LLA-B, general | 87,1 % ± 0,4 % (n = 7682) | <0,0001 | 59,1% ± 6,5% (n = 66) | 0,009 | 39,1% ± 7,9% (n = 40) | |
| LLA-B, RE | 90,8% ± 0,4 % (n = 5000) | 0,25 | 85,9% ± 7,6% (n = 22) | 0,45 | 76,2% ± 15,2% (n = 9) | |
| LLA-B, RA | 80% ± 0,9% (n = 2682) | <0,0001 | 44,9% ± 8,3% (n = 44) | 0,05 | 29% ± 8,2% (n = 31) | |
| LLA-T | 87,6% ± 1,5% (n = 1303) | 0,01 | 80,3% ± 7,3% (n = 97) | 0,13 | 62,7% ± 13,5% (n = 40) | |
| Tasas de supervivencia general: | ||||||
| LLA-B, general | 93,8% ± 0,3% (n = 7682) | <0,0001 | 77,2% ± 5,6% (n = 66) | 0,01 | 59% ± 8,9% (n = 40) | |
| LLA-B, RE | 96,6% ± 0,3% (n = 5000) | 0,24 | 95,5 % ± 4,6 % (n = 22) | 0,75 | 88,9% ± 12,1% (n = 9) | |
| LLA-B, RA | 88,4% ± 0,7% (n = 2682) | <0,0001 | 66,9% ± 8,3% (n = 44) | 0,06 | 51,4% ± 10,4% (n = 31) | |
| LLA-T | 91,9% ± 1,3% (n = 1303) | 0,005 | 83,4% ± 6,8% (n = 97) | 0,34 | 76,7% ± 12,3% (n = 40) | |
Grupos pronósticos (riesgo)
Durante décadas, los grupos de ensayos clínicos que estudian la LLA infantil han usado esquemas de clasificación del riesgo para asignar a los pacientes a los regímenes terapéuticos, de acuerdo con el cálculo del riesgo de fracaso del tratamiento. En los primeros sistemas de clasificación de riesgo se usaban factores clínicos, como la edad y el recuento de GB, en el momento de la presentación inicial. Luego se añadió la respuesta a las medidas terapéuticas; algunos grupos usan la respuesta morfológica temprana en la médula ósea (por ejemplo, en el día 8 o 15) y otros grupos utilizan la respuesta de las células leucémicas circulantes a la monoterapia con prednisona. En los sistemas contemporáneos de clasificación del riesgo, se continúan usando factores clínicos como la edad y el recuento de GB en el momento de la presentación inicial y se incorporan las alteraciones citogenéticas y genómicas de las células leucémicas en el momento del diagnóstico (por ejemplo, translocaciones favorables y desfavorables) y la respuesta al tratamiento a partir de la detección de la ERM al final de la inducción (y en algunos casos, en momentos posteriores). A continuación, se describen de manera breve los sistemas de clasificación del riesgo de los grupos COG y BFM.
Grupos de riesgo del Children’s Oncology Group
En los protocolos del Children's Oncology Group (COG), al inicio se estratifica a los niños con LLA en grupos de tratamiento (con diferentes grados de riesgo de fracaso del tratamiento) a partir de un subconjunto de factores pronósticos, como los siguientes:
- Edad.
- Recuento de GB en el momento del diagnóstico.
- Inmunofenotipo.
- Alteraciones citogenéticas o genómicas.
- Presencia de enfermedad extramedular.
- Síndrome de Down.
- Pretratamiento con corticoesteroides.
Las tasas de SSC superan el 85 % en los niños que cumplen con los criterios de pronóstico favorable (1 a <10 años de edad, recuento de GB <50 000/μl e inmunofenotipo de células B precursoras). En los niños que cumplen con los criterios de riesgo alto, las tasas de SSC son de alrededor del 75 %. Otros factores, como las anomalías citogenéticas y las mediciones de la respuesta temprana al tratamiento (por ejemplo, el índice de ERM en la sangre periférica en el día 8 y en muestras de la médula ósea al final de la inducción) junto con la edad de presentación, el recuento de GB, el inmunofenotipo, la presencia de enfermedad extramedular y el tratamiento previo con corticoesteroides permiten identificar grupos de pacientes para la terapia de posinducción con tasas previstas de SSC que oscilan entre menos del 40 % y más del 95 %.
Los pacientes con un riesgo muy alto de fracaso terapéutico son los siguientes:
- Lactantes con reordenamientos de KMT2A.
- Pacientes con hipodiploidía (<44 cromosomas).
- Pacientes con fracaso de la inducción inicial.
Grupos de riesgo de Berlin-Frankfurt-Münster
Desde el año 2000, la estratificación de riesgo en los protocolos del grupo Berlin-Frankfurt-Münster (BFM) se hizo teniendo en cuenta los criterios de respuesta al tratamiento y las características biológicas. La respuesta al tratamiento se determina sobre todo mediante mediciones de la ERM en dos momentos: al final de la inducción (punto de referencia 1, semana 5) y al final de la fase IB (similar a la fase de consolidación del COG) en la semana 12 (punto de referencia 2). Una ERM alta en ambos puntos de referencia se define como mayor de 5 × 10-4.
El BFM define 3 grupos de riesgo en función de la respuesta temprana:
- Riesgo estándar: pacientes con ERM negativa en ambos puntos de referencia.
- Riesgo intermedio: pacientes con ERM alta en el punto de referencia 1 y ERM negativa en el punto de referencia 2.
- Riesgo alto: pacientes con ERM alta en el punto de referencia 2. Los pacientes con LLA-T y una respuesta precaria a la profase de prednisona también se consideran de riesgo alto, con independencia de la ERM posterior.
Entre las características biológicas para estratificar a los pacientes como de riesgo alto (con independencia de la ERM en cualquiera de los puntos de referencia) se incluyen KMT2A::AFF1, TCF3::HLF e hipodiploidía (<45 cromosomas). Los pacientes con un estado IKZF1 plus (deleciones de IKZF1 que se presentan junto con deleciones de CDKN2A, CDKN2B, PAX5 o PAR1 en ausencia de deleción de ERG) se consideran de riesgo alto si presentan ERM al final de la inducción, independientemente de la ERM al final de la consolidación. La edad, el recuento leucocitario inicial y el estado del SNC en el momento del diagnóstico no se toman en consideración en el modelo actual de clasificación del riesgo.
Grupos pronósticos (riesgo) en evaluación clínica
- Ensayos clínicos del COG AALL1731 (NCT03914625) de riesgo estándar y AALL1732 (NCT03959085) de riesgo alto: el COG clasifica a los pacientes de LLA-B en 6 grupos de riesgo (riesgo estándar favorable, riesgo estándar promedio, riesgo estándar alto, riesgo alto favorable, riesgo alto y riesgo muy alto) a partir de las siguientes características:
- Edad y recuento leucocitario en el momento de la presentación inicial (criterios de los grupos de riesgo del NCI).
- Riesgo estándar (bajo) del NCI: incluye a los niños de 1 a <10 años con GB <50 000/µl en el momento del diagnóstico.
- Riesgo alto del NCI: incluye a los niños ≥10 años o con GB ≥ 50 000/µl en el momento del diagnóstico.
- Enfermedad extramedular (presencia o ausencia de leucemia en el SNC o los testículos).
- SNC1: ausencia de blastocitos en la preparación del LCR para la citocentrífuga, sin importar el número de GB.
- SNC2: presencia de <5 GB/μl en el LCR y resultado positivo para blastocitos en la prueba con citocentrífuga; o PL traumática, ≥5 GB/μl, resultado positivo para blastocitos en la prueba con citocentrífuga, pero un resultado negativo en el algoritmo de Steinherz/Bleyer.
- El grupo SNC3 se divide en los siguientes tipos:
- SNC3a: <10 GR/μl; ≥5 GR/μl y resultado positivo para blastocitos en la prueba con citocentrífuga.
- SNC3b: ≥ 10 GR/μl; ≥5 GR/μl y resultado positivo en el algoritmo de Steinherz/Bleyer.
- SNC3c: signos clínicos de leucemia en el SNC (parálisis del nervio facial, compromiso ocular o encefálico o síndrome hipotalámico).
- Alteraciones genómicas en las células leucémicas.
- Presencia de ERM en la sangre periférica en el día 8.
- Presencia de ERM y respuesta morfológica en la médula ósea en el día 29.
- Presencia de ERM al final de la consolidación.
- Pretratamiento con corticoesteroides.
En la actualidad ya no se realiza la evaluación morfológica de la respuesta temprana en la médula ósea en los días 8 y 15 de la inducción como parte de la estratificación del riesgo. Los pacientes con fenotipo de células T se tratan en ensayos separados y el riesgo no se clasifica de esta forma.
Las definiciones de características citogenéticas favorables, desfavorables y neutras para los pacientes con LLA-B se describen a continuación.
- Las características citogenéticas favorables son las siguientes:
- Hiperdiploidía con trisomía doble, en los cromosomas 4 y 10 (trisomía doble).
- Fusión ETV6::RUNX1.
- Las características citogenéticas desfavorables comprenden las siguientes:
- Hipodiploidía (<44 cromosomas o índice de DNA <0,81).
- Reordenamientos de KMT2A.
- t(17;19)(q21-q22;p13.3) o el transcrito resultante de la fusión TCF3::HLF.
- Amplificación intracromosómica del cromosoma 21 (iAMP21).
- Fusión BCR::ABL1 (Ph+ o t(9;22)(q34;q11)). Los pacientes con LLA positiva para BCR::ABL1 se tratan en un ensayo clínico separado.
- Características citogenéticas neutras: ausencia de características citogenéticas favorables o desfavorables.
Los pacientes de riesgo estándar del NCI se dividen en un grupo con desenlace muy favorable (riesgo estándar favorable; tasa de SSE a 5 años, >95 %), un grupo con desenlace favorable (riesgo estándar promedio; tasa de SSE a 5 años, 90–95 %), y el resto comprende el último grupo con una tasa de SSE a 5 años inferior al 90 % (riesgo estándar alto). Los pacientes del grupo de riesgo estándar alto reciben quimioterapia de posinducción de base según los regímenes para la LLA-B de riesgo alto con consolidación intensificada, mantenimiento provisional y terapia de reinducción. Los criterios para estos tres grupos se describen a continuación en el Cuadro 6, el Cuadro 7 y el Cuadro 8.
Cuadro 6. Leucemia linfoblástica aguda de células B de riesgo estándar favorable (con síndrome de Down o sin este)
Grupo de riesgo del NCI Estadio del SNC Pretratamiento con corticoesteroidesa Características genéticas favorables (ETV6::RUNX1 o TD) ERM en sangre periférica en el día 8 ERM en médula ósea en el día 29 SNC = sistema nervioso central; TD = trisomía doble; ERM = enfermedad residual mínima; NCI = Instituto Nacional del Cáncer; RE = riesgo estándar. aEn el mes previo al diagnóstico. RE 1, 2 Ausente Sí <1 % <0,01 % Cuadro 7. Leucemia linfoblástica aguda de células B de riesgo estándar promedio (con síndrome de Down y sin este)
Grupo de riesgo del NCI Estadio del SNC ETV6::RUNX1 TD Características citogenéticas neutras ERM en sangre periférica en el día 8 ERM en médula ósea en el día 29 SNC = sistema nervioso central; TD = trisomía doble; ERM = enfermedad residual mínima; NCI = Instituto Nacional del Cáncer; RE = riesgo estándar. RE 1, 2 Sí, cualquiera de los dos No ≥1 % <0,01 % RE 1, 2 No Sí No Cualquiera ≥0,01 % a <0,1 % RE 1 No No Sí Cualquiera <0,01 % Cuadro 8. Leucemia linfoblástica aguda de células B de riesgo estándar alto
Grupo de riesgo del NCI Estadio del SNC ETV6::RUNX1 TD Características citogenéticas neutras Características citogenéticas desfavorables ERM en sangre periférica en el día 8 ERM en médula ósea en el día 29 SNC = sistema nervioso central; TD = trisomía doble; ERM = enfermedad residual mínima; NCI = Instituto Nacional del Cáncer; RE = riesgo estándar. RE 1, 2 Sí No No No Cualquiera ≥0,01 % RE 1, 2 No Sí No No Cualquiera ≥0,1 % RE 1 No No Sí No Cualquiera ≥0,01 % RE 2 No No Sí No Cualquiera Cualquiera RE 1, 2 No No No Sí Cualquiera Cualquiera La LLA-B de riesgo alto favorable se define a partir de las características descritas en el Cuadro 9. Estos pacientes presentaron una tasa de SSC superior al 90 % en los ensayos clínicos del COG de pacientes con riesgo alto que se hicieron en el pasado.
Cuadro 9. Características de los pacientes con leucemia linfoblástica aguda de células B de riesgo alto favorable
Grupo de riesgo del NCI Edad (años) Estado del SNC Leucemia testicular Pretratamiento con corticoesteroides Características genéticas favorables (ETV6::RUNX1 o TD) ERM en médula ósea al final de la inducción RA <10 1 Ausente ≤24 horasa Sí <0,01 % SNC = sistema nervioso central; TD = trisomía doble; RA = riesgo alto; ERM = enfermedad residual mínima; NCI = Instituto Nacional del Cáncer. aDentro de las 2 semanas siguientes al diagnóstico. La LLA-B de riesgo alto se define a partir de las características descritas en el Cuadro 10. Los pacientes de riesgo estándar del NCI se pasan al grupo de riesgo alto a partir del pretratamiento con corticoesteroides y el compromiso en el SNC o en los testículos.
Cuadro 10. Características de los pacientes con leucemia linfoblástica aguda de células B de riesgo alto
Grupo de riesgo del NCI Edad (años) Leucemia en el SNC o testicular Pretratamiento con corticoesteroides Características citogenéticas ERM en médula ósea al final de la inducción ERM en médula ósea al final de la consolidación SNC = sistema nervioso central; RA = riesgo alto; ERM = enfermedad residual mínima; NA = no aplica; NCI = Instituto Nacional del Cáncer; RE = riesgo estándar. aSNC3. bSe excluye la LLA positiva para el cromosoma Filadelfia (Ph+). cSolo se evalúa la ERM en la médula ósea al final de la consolidación en los pacientes con ERM ≥0,01 % en la médula ósea al final de la inducción. dDentro de las 2 semanas siguientes al diagnóstico. eSNC2 o SNC3 RE <10 Sía Cualquiera Cualquierab Cualquiera <1 %c RE <10 No >24 horasd Cualquierab Cualquiera <1 %c RA ≥10 Cualquiera Cualquiera Cualquierab <0,01 % NA RA <10 Síe Cualquiera Cualquierab <0,01 % NA RA <10 No >24 horasd Cualquierab <0,01 % NA RA <10 No ≤24 horasd Neutro o desfavorableb <0,01 % NA RA Cualquiera Cualquiera Cualquiera Cualquierab ≥0,01 % <0,01 % Los pacientes de riesgo alto del NCI con ERM ≥0,01 % en la médula ósea al final de la consolidación se clasifican en el grupo de riesgo muy alto y son aptos para participar en un ensayo clínico de células T con receptor de antígeno quimérico (CAR) durante la primera remisión (NCT03792633).
Aunque los pacientes con LLA-B y síndrome de Down se clasifican en grupos de riesgo similares a otros niños, los pacientes con síndrome de Down del grupo de riesgo alto reciben un régimen de tratamiento modificado para disminuir la toxicidad.
- Edad y recuento leucocitario en el momento de la presentación inicial (criterios de los grupos de riesgo del NCI).
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
References
- Hunger SP, Loh ML, Whitlock JA, et al.: Children's Oncology Group's 2013 blueprint for research: acute lymphoblastic leukemia. Pediatr Blood Cancer 60 (6): 957-63, 2013.
- Hunger SP, Mullighan CG: Acute Lymphoblastic Leukemia in Children. N Engl J Med 373 (16): 1541-52, 2015.
- Smith M, Arthur D, Camitta B, et al.: Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol 14 (1): 18-24, 1996.
- Schultz KR, Pullen DJ, Sather HN, et al.: Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children's Cancer Group (CCG). Blood 109 (3): 926-35, 2007.
- Jeha S, Coustan-Smith E, Pei D, et al.: Impact of tyrosine kinase inhibitors on minimal residual disease and outcome in childhood Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer 120 (10): 1514-9, 2014.
- Vrooman LM, Silverman LB: Childhood acute lymphoblastic leukemia: update on prognostic factors. Curr Opin Pediatr 21 (1): 1-8, 2009.
- Möricke A, Zimmermann M, Reiter A, et al.: Prognostic impact of age in children and adolescents with acute lymphoblastic leukemia: data from the trials ALL-BFM 86, 90, and 95. Klin Padiatr 217 (6): 310-20, 2005 Nov-Dec.
- Vrooman LM, Blonquist TM, Harris MH, et al.: Refining risk classification in childhood B acute lymphoblastic leukemia: results of DFCI ALL Consortium Protocol 05-001. Blood Adv 2 (12): 1449-1458, 2018.
- Reaman GH, Sposto R, Sensel MG, et al.: Treatment outcome and prognostic factors for infants with acute lymphoblastic leukemia treated on two consecutive trials of the Children's Cancer Group. J Clin Oncol 17 (2): 445-55, 1999.
- Pieters R, Schrappe M, De Lorenzo P, et al.: A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370 (9583): 240-50, 2007.
- Hilden JM, Dinndorf PA, Meerbaum SO, et al.: Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children's Oncology Group. Blood 108 (2): 441-51, 2006.
- Dreyer ZE, Hilden JM, Jones TL, et al.: Intensified chemotherapy without SCT in infant ALL: results from COG P9407 (Cohort 3). Pediatr Blood Cancer 62 (3): 419-26, 2015.
- Chessells JM, Harrison CJ, Watson SL, et al.: Treatment of infants with lymphoblastic leukaemia: results of the UK Infant Protocols 1987-1999. Br J Haematol 117 (2): 306-14, 2002.
- Isoyama K, Eguchi M, Hibi S, et al.: Risk-directed treatment of infant acute lymphoblastic leukaemia based on early assessment of MLL gene status: results of the Japan Infant Leukaemia Study (MLL96). Br J Haematol 118 (4): 999-1010, 2002.
- Nagayama J, Tomizawa D, Koh K, et al.: Infants with acute lymphoblastic leukemia and a germline MLL gene are highly curable with use of chemotherapy alone: results from the Japan Infant Leukemia Study Group. Blood 107 (12): 4663-5, 2006.
- Sam TN, Kersey JH, Linabery AM, et al.: MLL gene rearrangements in infant leukemia vary with age at diagnosis and selected demographic factors: a Children's Oncology Group (COG) study. Pediatr Blood Cancer 58 (6): 836-9, 2012.
- Stam RW, Schneider P, de Lorenzo P, et al.: Prognostic significance of high-level FLT3 expression in MLL-rearranged infant acute lymphoblastic leukemia. Blood 110 (7): 2774-5, 2007.
- De Lorenzo P, Moorman AV, Pieters R, et al.: Cytogenetics and outcome of infants with acute lymphoblastic leukemia and absence of MLL rearrangements. Leukemia 28 (2): 428-30, 2014.
- Kang H, Wilson CS, Harvey RC, et al.: Gene expression profiles predictive of outcome and age in infant acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 119 (8): 1872-81, 2012.
- Andersson AK, Ma J, Wang J, et al.: The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet 47 (4): 330-7, 2015.
- Schrappe M, Reiter A, Ludwig WD, et al.: Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 95 (11): 3310-22, 2000.
- Vora A, Goulden N, Wade R, et al.: Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol 14 (3): 199-209, 2013.
- Place AE, Stevenson KE, Vrooman LM, et al.: Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): a randomised, open-label phase 3 trial. Lancet Oncol 16 (16): 1677-90, 2015.
- Forestier E, Schmiegelow K; on behalf of the Nordic Society of Paediatric Haematology and Oncology NOPHO: The incidence peaks of the childhood acute leukemias reflect specific cytogenetic aberrations. J Pediatr Hematol Oncol 28 (8): 486-95, 2006.
- Dastugue N, Suciu S, Plat G, et al.: Hyperdiploidy with 58-66 chromosomes in childhood B-acute lymphoblastic leukemia is highly curable: 58951 CLG-EORTC results. Blood 121 (13): 2415-23, 2013.
- Burke MJ, Devidas M, Chen Z, et al.: Outcomes in adolescent and young adult patients (16 to 30 years) compared to younger patients treated for high-risk B-lymphoblastic leukemia: report from Children's Oncology Group Study AALL0232. Leukemia 36 (3): 648-655, 2022.
- Nachman JB, La MK, Hunger SP, et al.: Young adults with acute lymphoblastic leukemia have an excellent outcome with chemotherapy alone and benefit from intensive postinduction treatment: a report from the children's oncology group. J Clin Oncol 27 (31): 5189-94, 2009.
- Pulte D, Gondos A, Brenner H: Improvement in survival in younger patients with acute lymphoblastic leukemia from the 1980s to the early 21st century. Blood 113 (7): 1408-11, 2009.
- Pui CH, Pei D, Campana D, et al.: Improved prognosis for older adolescents with acute lymphoblastic leukemia. J Clin Oncol 29 (4): 386-91, 2011.
- Childhood cancer. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 28. Also available online. Last accessed August 21, 2023.
- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 29. Also available online. Last accessed August 21, 2023.
- Smith MA, Ries LA, Gurney JG, et al.: Leukemia. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 17-34. Also available online. Last accessed August 11, 2022.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 15, 2023.
- de Bont JM, Holt B, Dekker AW, et al.: Significant difference in outcome for adolescents with acute lymphoblastic leukemia treated on pediatric vs adult protocols in the Netherlands. Leukemia 18 (12): 2032-5, 2004.
- Boissel N, Auclerc MF, Lhéritier V, et al.: Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-93 and LALA-94 trials. J Clin Oncol 21 (5): 774-80, 2003.
- Stock W, La M, Sanford B, et al.: What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children's Cancer Group and Cancer and Leukemia Group B studies. Blood 112 (5): 1646-54, 2008.
- Hastings C, Gaynon PS, Nachman JB, et al.: Increased post-induction intensification improves outcome in children and adolescents with a markedly elevated white blood cell count (≥200 × 10(9) /l) with T cell acute lymphoblastic leukaemia but not B cell disease: a report from the Children's Oncology Group. Br J Haematol 168 (4): 533-46, 2015.
- Pullen J, Shuster JJ, Link M, et al.: Significance of commonly used prognostic factors differs for children with T cell acute lymphocytic leukemia (ALL), as compared to those with B-precursor ALL. A Pediatric Oncology Group (POG) study. Leukemia 13 (11): 1696-707, 1999.
- Goldberg JM, Silverman LB, Levy DE, et al.: Childhood T-cell acute lymphoblastic leukemia: the Dana-Farber Cancer Institute acute lymphoblastic leukemia consortium experience. J Clin Oncol 21 (19): 3616-22, 2003.
- Silverman LB, Stevenson KE, O'Brien JE, et al.: Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia 24 (2): 320-34, 2010.
- Pui CH, Pei D, Sandlund JT, et al.: Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 24 (2): 371-82, 2010.
- Gaynon PS, Angiolillo AL, Carroll WL, et al.: Long-term results of the children's cancer group studies for childhood acute lymphoblastic leukemia 1983-2002: a Children's Oncology Group Report. Leukemia 24 (2): 285-97, 2010.
- Möricke A, Zimmermann M, Reiter A, et al.: Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 24 (2): 265-84, 2010.
- Vaitkevičienė G, Forestier E, Hellebostad M, et al.: High white blood cell count at diagnosis of childhood acute lymphoblastic leukaemia: biological background and prognostic impact. Results from the NOPHO ALL-92 and ALL-2000 studies. Eur J Haematol 86 (1): 38-46, 2011.
- Burns MA, Place AE, Stevenson KE, et al.: Identification of prognostic factors in childhood T-cell acute lymphoblastic leukemia: Results from DFCI ALL Consortium Protocols 05-001 and 11-001. Pediatr Blood Cancer 68 (1): e28719, 2021.
- Bürger B, Zimmermann M, Mann G, et al.: Diagnostic cerebrospinal fluid examination in children with acute lymphoblastic leukemia: significance of low leukocyte counts with blasts or traumatic lumbar puncture. J Clin Oncol 21 (2): 184-8, 2003.
- Vora A, Andreano A, Pui CH, et al.: Influence of Cranial Radiotherapy on Outcome in Children With Acute Lymphoblastic Leukemia Treated With Contemporary Therapy. J Clin Oncol 34 (9): 919-26, 2016.
- Gossai NP, Devidas M, Chen Z, et al.: Central nervous system status is prognostic in T-cell acute lymphoblastic leukemia: a Children's Oncology Group report. Blood 141 (15): 1802-1811, 2023.
- Mahmoud HH, Rivera GK, Hancock ML, et al.: Low leukocyte counts with blast cells in cerebrospinal fluid of children with newly diagnosed acute lymphoblastic leukemia. N Engl J Med 329 (5): 314-9, 1993.
- Winick N, Devidas M, Chen S, et al.: Impact of Initial CSF Findings on Outcome Among Patients With National Cancer Institute Standard- and High-Risk B-Cell Acute Lymphoblastic Leukemia: A Report From the Children's Oncology Group. J Clin Oncol 35 (22): 2527-2534, 2017.
- Sirvent N, Suciu S, Rialland X, et al.: Prognostic significance of the initial cerebro-spinal fluid (CSF) involvement of children with acute lymphoblastic leukaemia (ALL) treated without cranial irradiation: results of European Organization for Research and Treatment of Cancer (EORTC) Children Leukemia Group study 58881. Eur J Cancer 47 (2): 239-47, 2011.
- te Loo DM, Kamps WA, van der Does-van den Berg A, et al.: Prognostic significance of blasts in the cerebrospinal fluid without pleiocytosis or a traumatic lumbar puncture in children with acute lymphoblastic leukemia: experience of the Dutch Childhood Oncology Group. J Clin Oncol 24 (15): 2332-6, 2006.
- Gilchrist GS, Tubergen DG, Sather HN, et al.: Low numbers of CSF blasts at diagnosis do not predict for the development of CNS leukemia in children with intermediate-risk acute lymphoblastic leukemia: a Childrens Cancer Group report. J Clin Oncol 12 (12): 2594-600, 1994.
- Gajjar A, Harrison PL, Sandlund JT, et al.: Traumatic lumbar puncture at diagnosis adversely affects outcome in childhood acute lymphoblastic leukemia. Blood 96 (10): 3381-4, 2000.
- Matloub Y, Bostrom BC, Hunger SP, et al.: Escalating intravenous methotrexate improves event-free survival in children with standard-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 118 (2): 243-51, 2011.
- Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.
- Levinsen M, Taskinen M, Abrahamsson J, et al.: Clinical features and early treatment response of central nervous system involvement in childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 61 (8): 1416-21, 2014.
- Jeha S, Pei D, Choi J, et al.: Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J Clin Oncol 37 (35): 3377-3391, 2019.
- Cherlow JM, Sather H, Steinherz P, et al.: Craniospinal irradiation for acute lymphoblastic leukemia with central nervous system disease at diagnosis: a report from the Children's Cancer Group. Int J Radiat Oncol Biol Phys 36 (1): 19-27, 1996.
- Hijiya N, Liu W, Sandlund JT, et al.: Overt testicular disease at diagnosis of childhood acute lymphoblastic leukemia: lack of therapeutic role of local irradiation. Leukemia 19 (8): 1399-403, 2005.
- Sirvent N, Suciu S, Bertrand Y, et al.: Overt testicular disease (OTD) at diagnosis is not associated with a poor prognosis in childhood acute lymphoblastic leukemia: results of the EORTC CLG Study 58881. Pediatr Blood Cancer 49 (3): 344-8, 2007.
- Bassal M, La MK, Whitlock JA, et al.: Lymphoblast biology and outcome among children with Down syndrome and ALL treated on CCG-1952. Pediatr Blood Cancer 44 (1): 21-8, 2005.
- Zeller B, Gustafsson G, Forestier E, et al.: Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br J Haematol 128 (6): 797-804, 2005.
- Whitlock JA, Sather HN, Gaynon P, et al.: Clinical characteristics and outcome of children with Down syndrome and acute lymphoblastic leukemia: a Children's Cancer Group study. Blood 106 (13): 4043-9, 2005.
- Arico M, Ziino O, Valsecchi MG, et al.: Acute lymphoblastic leukemia and Down syndrome: presenting features and treatment outcome in the experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP). Cancer 113 (3): 515-21, 2008.
- Lundin C, Forestier E, Klarskov Andersen M, et al.: Clinical and genetic features of pediatric acute lymphoblastic leukemia in Down syndrome in the Nordic countries. J Hematol Oncol 7 (1): 32, 2014.
- Athale UH, Puligandla M, Stevenson KE, et al.: Outcome of children and adolescents with Down syndrome treated on Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium protocols 00-001 and 05-001. Pediatr Blood Cancer 65 (10): e27256, 2018.
- Matloub Y, Rabin KR, Ji L, et al.: Excellent long-term survival of children with Down syndrome and standard-risk ALL: a report from the Children's Oncology Group. Blood Adv 3 (11): 1647-1656, 2019.
- Maloney KW, Carroll WL, Carroll AJ, et al.: Down syndrome childhood acute lymphoblastic leukemia has a unique spectrum of sentinel cytogenetic lesions that influences treatment outcome: a report from the Children's Oncology Group. Blood 116 (7): 1045-50, 2010.
- Buitenkamp TD, Izraeli S, Zimmermann M, et al.: Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood 123 (1): 70-7, 2014.
- Li Z, Chang TC, Junco JJ, et al.: Genomic landscape of Down syndrome-associated acute lymphoblastic leukemia. Blood 142 (2): 172-184, 2023.
- Mullighan CG, Collins-Underwood JR, Phillips LA, et al.: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243-6, 2009.
- Bercovich D, Ganmore I, Scott LM, et al.: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484-92, 2008.
- Gaikwad A, Rye CL, Devidas M, et al.: Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol 144 (6): 930-2, 2009.
- Kearney L, Gonzalez De Castro D, Yeung J, et al.: Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood 113 (3): 646-8, 2009.
- Buitenkamp TD, Pieters R, Gallimore NE, et al.: Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia 26 (10): 2204-11, 2012.
- Hanada I, Terui K, Ikeda F, et al.: Gene alterations involving the CRLF2-JAK pathway and recurrent gene deletions in Down syndrome-associated acute lymphoblastic leukemia in Japan. Genes Chromosomes Cancer 53 (11): 902-10, 2014.
- Pui CH, Boyett JM, Relling MV, et al.: Sex differences in prognosis for children with acute lymphoblastic leukemia. J Clin Oncol 17 (3): 818-24, 1999.
- Shuster JJ, Wacker P, Pullen J, et al.: Prognostic significance of sex in childhood B-precursor acute lymphoblastic leukemia: a Pediatric Oncology Group Study. J Clin Oncol 16 (8): 2854-63, 1998.
- Chessells JM, Richards SM, Bailey CC, et al.: Gender and treatment outcome in childhood lymphoblastic leukaemia: report from the MRC UKALL trials. Br J Haematol 89 (2): 364-72, 1995.
- Silverman LB, Gelber RD, Dalton VK, et al.: Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood 97 (5): 1211-8, 2001.
- Hunger SP, Lu X, Devidas M, et al.: Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol 30 (14): 1663-9, 2012.
- Gupta S, Teachey DT, Chen Z, et al.: Sex-based disparities in outcome in pediatric acute lymphoblastic leukemia: a Children's Oncology Group report. Cancer 128 (9): 1863-1870, 2022.
- Bhatia S: Influence of race and socioeconomic status on outcome of children treated for childhood acute lymphoblastic leukemia. Curr Opin Pediatr 16 (1): 9-14, 2004.
- Kadan-Lottick NS, Ness KK, Bhatia S, et al.: Survival variability by race and ethnicity in childhood acute lymphoblastic leukemia. JAMA 290 (15): 2008-14, 2003.
- Tai EW, Ward KC, Bonaventure A, et al.: Survival among children diagnosed with acute lymphoblastic leukemia in the United States, by race and age, 2001 to 2009: Findings from the CONCORD-2 study. Cancer 123 (Suppl 24): 5178-5189, 2017.
- Kahn JM, Cole PD, Blonquist TM, et al.: An investigation of toxicities and survival in Hispanic children and adolescents with ALL: Results from the Dana-Farber Cancer Institute ALL Consortium protocol 05-001. Pediatr Blood Cancer 65 (3): , 2018.
- Gupta S, Dai Y, Chen Z, et al.: Racial and ethnic disparities in childhood and young adult acute lymphocytic leukaemia: secondary analyses of eight Children's Oncology Group cohort trials. Lancet Haematol 10 (2): e129-e141, 2023.
- Lee SHR, Antillon-Klussmann F, Pei D, et al.: Association of Genetic Ancestry With the Molecular Subtypes and Prognosis of Childhood Acute Lymphoblastic Leukemia. JAMA Oncol 8 (3): 354-363, 2022.
- Harvey RC, Mullighan CG, Chen IM, et al.: Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 115 (26): 5312-21, 2010.
- Raca G, Abdel-Azim H, Yue F, et al.: Increased Incidence of IKZF1 deletions and IGH-CRLF2 translocations in B-ALL of Hispanic/Latino children-a novel health disparity. Leukemia 35 (8): 2399-2402, 2021.
- Bhatia S, Landier W, Shangguan M, et al.: Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the children's oncology group. J Clin Oncol 30 (17): 2094-101, 2012.
- Bhatia S, Landier W, Hageman L, et al.: 6MP adherence in a multiracial cohort of children with acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 124 (15): 2345-53, 2014.
- Yang JJ, Cheng C, Devidas M, et al.: Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet 43 (3): 237-41, 2011.
- Xu H, Cheng C, Devidas M, et al.: ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J Clin Oncol 30 (7): 751-7, 2012.
- Perez-Andreu V, Roberts KG, Harvey RC, et al.: Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet 45 (12): 1494-8, 2013.
- Aldhafiri FK, McColl JH, Reilly JJ: Prognostic significance of being overweight and obese at diagnosis in children with acute lymphoblastic leukemia. J Pediatr Hematol Oncol 36 (3): 234-6, 2014.
- Baillargeon J, Langevin AM, Lewis M, et al.: Obesity and survival in a cohort of predominantly Hispanic children with acute lymphoblastic leukemia. J Pediatr Hematol Oncol 28 (9): 575-8, 2006.
- Hijiya N, Panetta JC, Zhou Y, et al.: Body mass index does not influence pharmacokinetics or outcome of treatment in children with acute lymphoblastic leukemia. Blood 108 (13): 3997-4002, 2006.
- Butturini AM, Dorey FJ, Lange BJ, et al.: Obesity and outcome in pediatric acute lymphoblastic leukemia. J Clin Oncol 25 (15): 2063-9, 2007.
- Gelelete CB, Pereira SH, Azevedo AM, et al.: Overweight as a prognostic factor in children with acute lymphoblastic leukemia. Obesity (Silver Spring) 19 (9): 1908-11, 2011.
- Orgel E, Sposto R, Malvar J, et al.: Impact on survival and toxicity by duration of weight extremes during treatment for pediatric acute lymphoblastic leukemia: A report from the Children's Oncology Group. J Clin Oncol 32 (13): 1331-7, 2014.
- Orgel E, Tucci J, Alhushki W, et al.: Obesity is associated with residual leukemia following induction therapy for childhood B-precursor acute lymphoblastic leukemia. Blood 124 (26): 3932-8, 2014.
- Eissa HM, Zhou Y, Panetta JC, et al.: The effect of body mass index at diagnosis on clinical outcome in children with newly diagnosed acute lymphoblastic leukemia. Blood Cancer J 7 (2): e531, 2017.
- Egnell C, Heyman M, Jónsson ÓG, et al.: Obesity as a predictor of treatment-related toxicity in children with acute lymphoblastic leukaemia. Br J Haematol 196 (5): 1239-1247, 2022.
- Shimony S, Flamand Y, Valtis YK, et al.: Effect of BMI on toxicities and survival among adolescents and young adults treated on DFCI Consortium ALL trials. Blood Adv 7 (18): 5234-5245, 2023.
- den Hoed MA, Pluijm SM, de Groot-Kruseman HA, et al.: The negative impact of being underweight and weight loss on survival of children with acute lymphoblastic leukemia. Haematologica 100 (1): 62-9, 2015.
- Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. International Agency for Research on Cancer, 2017.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Pui CH, Chessells JM, Camitta B, et al.: Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia 17 (4): 700-6, 2003.
- Möricke A, Ratei R, Ludwig WD, et al.: Prognostic factors in CD10 negative precursor b-cell acute lymphoblastic leukemia in children: data from three consecutive trials ALL-BFM 86, 90, and 95. [Abstract] Blood 104 (11): A-1957, 540a, 2004.
- Hunger SP: Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood 87 (4): 1211-24, 1996.
- Uckun FM, Sensel MG, Sather HN, et al.: Clinical significance of translocation t(1;19) in childhood acute lymphoblastic leukemia in the context of contemporary therapies: a report from the Children's Cancer Group. J Clin Oncol 16 (2): 527-35, 1998.
- Koehler M, Behm FG, Shuster J, et al.: Transitional pre-B-cell acute lymphoblastic leukemia of childhood is associated with favorable prognostic clinical features and an excellent outcome: a Pediatric Oncology Group study. Leukemia 7 (12): 2064-8, 1993.
- Wagener R, López C, Kleinheinz K, et al.: IG-MYC+ neoplasms with precursor B-cell phenotype are molecularly distinct from Burkitt lymphomas. Blood 132 (21): 2280-2285, 2018.
- Winter SS, Dunsmore KP, Devidas M, et al.: Improved Survival for Children and Young Adults With T-Lineage Acute Lymphoblastic Leukemia: Results From the Children's Oncology Group AALL0434 Methotrexate Randomization. J Clin Oncol 36 (29): 2926-2934, 2018.
- Slack JL, Arthur DC, Lawrence D, et al.: Secondary cytogenetic changes in acute promyelocytic leukemia--prognostic importance in patients treated with chemotherapy alone and association with the intron 3 breakpoint of the PML gene: a Cancer and Leukemia Group B study. J Clin Oncol 15 (5): 1786-95, 1997.
- Attarbaschi A, Mann G, Dworzak M, et al.: Mediastinal mass in childhood T-cell acute lymphoblastic leukemia: significance and therapy response. Med Pediatr Oncol 39 (6): 558-65, 2002.
- Coustan-Smith E, Mullighan CG, Onciu M, et al.: Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol 10 (2): 147-56, 2009.
- Ma M, Wang X, Tang J, et al.: Early T-cell precursor leukemia: a subtype of high risk childhood acute lymphoblastic leukemia. Front Med 6 (4): 416-20, 2012.
- Inukai T, Kiyokawa N, Campana D, et al.: Clinical significance of early T-cell precursor acute lymphoblastic leukaemia: results of the Tokyo Children's Cancer Study Group Study L99-15. Br J Haematol 156 (3): 358-65, 2012.
- Patrick K, Wade R, Goulden N, et al.: Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br J Haematol 166 (3): 421-4, 2014.
- Dunsmore KP, Winter SS, Devidas M, et al.: Children's Oncology Group AALL0434: A Phase III Randomized Clinical Trial Testing Nelarabine in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia. J Clin Oncol 38 (28): 3282-3293, 2020.
- Wood BL, Winter SS, Dunsmore KP, et al.: T-lymphoblastic leukemia (T-ALL) shows excellent outcome, lack of significance of the early thymic precursor (ETP) immunophenotype, and validation of the prognostic value of end-induction minimal residual disease (MRD) in Children’s Oncology Group (COG) study AALL0434. [Abstract] Blood 124 (21): A-1, 2014. Also available online. Last accessed June 04, 2021.
- Pui CH, Rubnitz JE, Hancock ML, et al.: Reappraisal of the clinical and biologic significance of myeloid-associated antigen expression in childhood acute lymphoblastic leukemia. J Clin Oncol 16 (12): 3768-73, 1998.
- Uckun FM, Sather HN, Gaynon PS, et al.: Clinical features and treatment outcome of children with myeloid antigen positive acute lymphoblastic leukemia: a report from the Children's Cancer Group. Blood 90 (1): 28-35, 1997.
- Corrente F, Bellesi S, Metafuni E, et al.: Role of flow-cytometric immunophenotyping in prediction of BCR/ABL1 gene rearrangement in adult B-cell acute lymphoblastic leukemia. Cytometry B Clin Cytom 94 (3): 468-476, 2018.
- Hirabayashi S, Ohki K, Nakabayashi K, et al.: ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 102 (1): 118-129, 2017.
- Qian M, Zhang H, Kham SK, et al.: Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res 27 (2): 185-195, 2017.
- Relling MV, Dervieux T: Pharmacogenetics and cancer therapy. Nat Rev Cancer 1 (2): 99-108, 2001.
- van Dongen JJ, Seriu T, Panzer-Grümayer ER, et al.: Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 352 (9142): 1731-8, 1998.
- Wood B, Wu D, Crossley B, et al.: Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood 131 (12): 1350-1359, 2018.
- Zhou J, Goldwasser MA, Li A, et al.: Quantitative analysis of minimal residual disease predicts relapse in children with B-lineage acute lymphoblastic leukemia in DFCI ALL Consortium Protocol 95-01. Blood 110 (5): 1607-11, 2007.
- Borowitz MJ, Devidas M, Hunger SP, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood 111 (12): 5477-85, 2008.
- Borowitz MJ, Wood BL, Devidas M, et al.: Prognostic significance of minimal residual disease in high risk B-ALL: a report from Children's Oncology Group study AALL0232. Blood 126 (8): 964-71, 2015.
- Conter V, Bartram CR, Valsecchi MG, et al.: Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 115 (16): 3206-14, 2010.
- O'Connor D, Enshaei A, Bartram J, et al.: Genotype-Specific Minimal Residual Disease Interpretation Improves Stratification in Pediatric Acute Lymphoblastic Leukemia. J Clin Oncol 36 (1): 34-43, 2018.
- Basso G, Veltroni M, Valsecchi MG, et al.: Risk of relapse of childhood acute lymphoblastic leukemia is predicted by flow cytometric measurement of residual disease on day 15 bone marrow. J Clin Oncol 27 (31): 5168-74, 2009.
- Pui CH, Pei D, Coustan-Smith E, et al.: Clinical utility of sequential minimal residual disease measurements in the context of risk-based therapy in childhood acute lymphoblastic leukaemia: a prospective study. Lancet Oncol 16 (4): 465-74, 2015.
- Rau RE, Dai Y, Devidas M, et al.: Prognostic impact of minimal residual disease at the end of consolidation in NCI standard-risk B-lymphoblastic leukemia: A report from the Children's Oncology Group. Pediatr Blood Cancer 68 (4): e28929, 2021.
- Schrappe M, Valsecchi MG, Bartram CR, et al.: Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood 118 (8): 2077-84, 2011.
- Teachey DT, Devidas M, Wood BL, et al.: Children's Oncology Group Trial AALL1231: A Phase III Clinical Trial Testing Bortezomib in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia and Lymphoma. J Clin Oncol 40 (19): 2106-2118, 2022.
- Bartram J, Wade R, Vora A, et al.: Excellent outcome of minimal residual disease-defined low-risk patients is sustained with more than 10 years follow-up: results of UK paediatric acute lymphoblastic leukaemia trials 1997-2003. Arch Dis Child 101 (5): 449-54, 2016.
- Vora A, Goulden N, Mitchell C, et al.: Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): a randomised controlled trial. Lancet Oncol 15 (8): 809-18, 2014.
- Pieters R, de Groot-Kruseman H, Van der Velden V, et al.: Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol 34 (22): 2591-601, 2016.
- Gaynon PS, Desai AA, Bostrom BC, et al.: Early response to therapy and outcome in childhood acute lymphoblastic leukemia: a review. Cancer 80 (9): 1717-26, 1997.
- Borowitz MJ, Wood BL, Devidas M, et al.: Assessment of end induction minimal residual disease (MRD) in childhood B precursor acute lymphoblastic leukemia (ALL) to eliminate the need for day 14 marrow examination: A Children’s Oncology Group study. [Abstract] J Clin Oncol 31 (Suppl 15): A-10001, 2013. Also available online. Last accessed June 04, 2021.
- Möricke A, Reiter A, Zimmermann M, et al.: Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 111 (9): 4477-89, 2008.
- Griffin TC, Shuster JJ, Buchanan GR, et al.: Slow disappearance of peripheral blood blasts is an adverse prognostic factor in childhood T cell acute lymphoblastic leukemia: a Pediatric Oncology Group study. Leukemia 14 (5): 792-5, 2000.
- Volejnikova J, Mejstrikova E, Valova T, et al.: Minimal residual disease in peripheral blood at day 15 identifies a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with superior prognosis. Haematologica 96 (12): 1815-21, 2011.
- Schrappe M, Hunger SP, Pui CH, et al.: Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med 366 (15): 1371-81, 2012.
- Möricke A, Zimmermann M, Valsecchi MG, et al.: Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood 127 (17): 2101-12, 2016.
- O'Connor D, Moorman AV, Wade R, et al.: Use of Minimal Residual Disease Assessment to Redefine Induction Failure in Pediatric Acute Lymphoblastic Leukemia. J Clin Oncol 35 (6): 660-667, 2017.
- Silverman LB, Gelber RD, Young ML, et al.: Induction failure in acute lymphoblastic leukemia of childhood. Cancer 85 (6): 1395-404, 1999.
- Oudot C, Auclerc MF, Levy V, et al.: Prognostic factors for leukemic induction failure in children with acute lymphoblastic leukemia and outcome after salvage therapy: the FRALLE 93 study. J Clin Oncol 26 (9): 1496-503, 2008.
- Schwab C, Ryan SL, Chilton L, et al.: EBF1-PDGFRB fusion in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL): genetic profile and clinical implications. Blood 127 (18): 2214-8, 2016.
- den Boer ML, Cario G, Moorman AV, et al.: Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: a multicentre, retrospective, cohort study. Lancet Haematol 8 (1): e55-e66, 2021.
- Gupta S, Devidas M, Loh ML, et al.: Flow-cytometric vs. -morphologic assessment of remission in childhood acute lymphoblastic leukemia: a report from the Children's Oncology Group (COG). Leukemia 32 (6): 1370-1379, 2018.
- Moghrabi A, Levy DE, Asselin B, et al.: Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood 109 (3): 896-904, 2007.
- Veerman AJ, Kamps WA, van den Berg H, et al.: Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004). Lancet Oncol 10 (10): 957-66, 2009.
- Kosaka Y, Koh K, Kinukawa N, et al.: Infant acute lymphoblastic leukemia with MLL gene rearrangements: outcome following intensive chemotherapy and hematopoietic stem cell transplantation. Blood 104 (12): 3527-34, 2004.
- Balduzzi A, Valsecchi MG, Uderzo C, et al.: Chemotherapy versus allogeneic transplantation for very-high-risk childhood acute lymphoblastic leukaemia in first complete remission: comparison by genetic randomisation in an international prospective study. Lancet 366 (9486): 635-42, 2005 Aug 20-26.
- Schrauder A, Reiter A, Gadner H, et al.: Superiority of allogeneic hematopoietic stem-cell transplantation compared with chemotherapy alone in high-risk childhood T-cell acute lymphoblastic leukemia: results from ALL-BFM 90 and 95. J Clin Oncol 24 (36): 5742-9, 2006.
- Ribera JM, Ortega JJ, Oriol A, et al.: Comparison of intensive chemotherapy, allogeneic, or autologous stem-cell transplantation as postremission treatment for children with very high risk acute lymphoblastic leukemia: PETHEMA ALL-93 Trial. J Clin Oncol 25 (1): 16-24, 2007.
- Stanulla M, Dagdan E, Zaliova M, et al.: IKZF1plus Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J Clin Oncol 36 (12): 1240-1249, 2018.
Aspectos generales de las opciones de tratamiento de la leucemia linfoblástica aguda infantil
Fases del tratamiento
El tratamiento de los niños con leucemia linfoblástica aguda (LLA) por lo general se divide en las siguientes fases:
- Quimioterapia de inducción a la remisión (en el momento del diagnóstico).
- Terapia de posinducción (después de alcanzar la remisión completa).
- Terapia de consolidación o intensificación.
- Terapia de mantenimiento.
Sitios santuario
En el pasado, determinados sitios extramedulares se consideraban sitios santuario (es decir, espacios anatómicos en los que muchos de los fármacos quimioterapéuticos de administración oral o intravenosa que a menudo se usan para el tratamiento de la LLA tienen poca penetración). Los dos sitios santuario más importantes en la LLA infantil son el sistema nervioso central (SNC) y los testículos. El tratamiento exitoso de la LLA requiere un abordaje eficaz del compromiso leucémico clínico o subclínico en estos sitios considerados santuarios extramedulares.
Sistema nervioso central
En el momento del diagnóstico, cerca del 3 % de los pacientes tienen enfermedad SNC3 (definida por una muestra de líquido cefalorraquídeo con ≥5 glóbulos blancos/μl, linfoblastos o parálisis de nervios craneales). Sin embargo, a menos que se dirija una terapia específica al sistema nervioso central (SNC), la mayoría de los niños presentará con el tiempo leucemia en el SNC evidente, sin importar si se detectaron linfoblastos o no en el líquido cefalorraquídeo (LCR) en el momento del diagnóstico inicial. Los tratamientos dirigidos al SNC incluyen la quimioterapia intratecal, la quimioterapia sistémica dirigida al SNC y la radiación craneal. Algunos o todos estos tratamientos se incluyen en los regímenes que se usan en la actualidad para la LLA. Para obtener más información, consultar la sección Terapia dirigida al sistema nervioso central para la leucemia linfoblástica aguda infantil.
Testículos
El compromiso testicular evidente en el momento del diagnóstico se presenta en cerca del 2 % de los varones. En los primeros ensayos de la LLA, el compromiso testicular en el momento del diagnóstico era un factor de pronóstico adverso. Sin embargo, no está clara la importancia pronóstica del compromiso testicular inicial cuando se usa un tratamiento inicial más intensivo. Tampoco está clara la función de la radioterapia para el compromiso testicular. En un estudio del St. Jude Children's Research Hospital, se indicó que se logra un desenlace favorable cuando se usa quimioterapia convencional intensiva sin radiación. El Children's Oncology Group también adoptó esta estrategia para los niños con compromiso testicular que se resuelve por completo durante la quimioterapia de inducción.
References
- Hijiya N, Liu W, Sandlund JT, et al.: Overt testicular disease at diagnosis of childhood acute lymphoblastic leukemia: lack of therapeutic role of local irradiation. Leukemia 19 (8): 1399-403, 2005.
- Sirvent N, Suciu S, Bertrand Y, et al.: Overt testicular disease (OTD) at diagnosis is not associated with a poor prognosis in childhood acute lymphoblastic leukemia: results of the EORTC CLG Study 58881. Pediatr Blood Cancer 49 (3): 344-8, 2007.
Consideraciones especiales para el tratamiento de niños con leucemia linfoblástica aguda
La evaluación y el tratamiento de niños y adolescentes con leucemia linfoblástica aguda (LLA) supone una compleja evaluación del riesgo, tratamientos extensos y cuidados médicos de apoyo intensivos (por ejemplo, transfusiones y tratamiento de complicaciones infecciosas, además de apoyo emocional, financiero y del desarrollo). Por todo esto, la evaluación y el tratamiento de estos pacientes debe estar coordinado por un equipo multidisciplinario en centros oncológicos u hospitales que cuenten con todas las instalaciones pediátricas de apoyo. Este equipo multidisciplinario incorpora la pericia de los siguientes especialistas en pediatría y otros para asegurar que los niños reciban el tratamiento, los cuidados médicos de apoyo y la rehabilitación que les permitan lograr una supervivencia y calidad de vida óptimas:
- Médicos de atención primaria.
- Oncólogos y hematólogos pediatras.
- Cirujanos pediatras.
- Patólogos.
- Radioncólogos pediatras.
- Pediatras intensivistas.
- Especialistas en rehabilitación.
- Enfermeros de oncología pediátrica.
- Trabajadores sociales.
- Profesionales de la vida infantil.
- Psicólogos.
- Nutricionistas y dietistas.
Para obtener información específica sobre los cuidados médicos de apoyo para niños y adolescentes con cáncer, consultar los resúmenes de Cuidados médicos de apoyo y cuidados paliativos.
La American Academy of Pediatrics estableció pautas para los centros de oncología pediátrica y su función en el tratamiento de los niños y adolescentes con cáncer. El tratamiento de la LLA infantil a menudo incluye quimioterapia durante 2 a 3 años. Debido a que la mielodepresión y la inmunodepresión generalizada son consecuencias anticipadas de la leucemia y la quimioterapia, se debe disponer de forma inmediata de instalaciones adecuadas para el apoyo hematológico y el tratamiento de infecciones y otras complicaciones durante todas las fases del tratamiento. Alrededor del 1 % al 3 % de los pacientes muere durante la fase de inducción a la remisión y otro 1 % al 3 % muere después de la remisión completa por complicaciones relacionadas con el tratamiento. Es importante que los centros asistenciales y los especialistas encargados de la atención del paciente mantengan contacto con el médico de atención primaria que lo derivó. Una comunicación sólida optimiza cualquier atención de urgencia o cuidados provisionales que necesite el niño en su hogar.
Por lo general, hay ensayos clínicos para los niños con LLA en los que se usan protocolos específicos diseñados para los niños con un riesgo estándar (bajo) de fracaso del tratamiento y para los niños con un riesgo más alto de fracaso del tratamiento. Los ensayos clínicos para niños con LLA a menudo se diseñan a fin de comparar el tratamiento estándar para un grupo de riesgo en particular con un abordaje de tratamiento que quizás mejore la supervivencia o reduzca la toxicidad relacionada con el régimen de tratamiento estándar. En otros tipos de ensayos clínicos se prueban tratamientos novedosos cuando no hay un tratamiento estándar para el cáncer que se ha diagnosticado. La mayoría de las innovaciones terapéuticas que aumentaron las tasas de supervivencia de los niños con LLA se lograron mediante ensayos clínicos, por lo tanto es conveniente que a los niños y adolescentes con LLA se les ofrezca la participación en un ensayo clínico. Para obtener información sobre los ensayos clínicos en curso, consultar el portal de Internet del NCI.
La asignación del tratamiento según el riesgo es una estrategia terapéutica importante en los niños con LLA. Con este abordaje, los niños que en el pasado obtuvieron resultados muy favorables reciben un tratamiento menos intensivo y evitan tratamientos más tóxicos, mientras que los niños con una probabilidad más baja de sobrevivir a largo plazo reciben un tratamiento más intensivo que aumenta la probabilidad de curación. Para obtener más información acerca de las manifestaciones clínicas y de laboratorio con importancia pronóstica, consultar la sección Asignación del tratamiento según el riesgo.
References
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed December 15, 2023.
- Rubnitz JE, Lensing S, Zhou Y, et al.: Death during induction therapy and first remission of acute leukemia in childhood: the St. Jude experience. Cancer 101 (7): 1677-84, 2004.
- Christensen MS, Heyman M, Möttönen M, et al.: Treatment-related death in childhood acute lymphoblastic leukaemia in the Nordic countries: 1992-2001. Br J Haematol 131 (1): 50-8, 2005.
- Vrooman LM, Stevenson KE, Supko JG, et al.: Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol 31 (9): 1202-10, 2013.
- Lund B, Åsberg A, Heyman M, et al.: Risk factors for treatment related mortality in childhood acute lymphoblastic leukaemia. Pediatr Blood Cancer 56 (4): 551-9, 2011.
- Alvarez EM, Malogolowkin M, Li Q, et al.: Decreased Early Mortality in Young Adult Patients With Acute Lymphoblastic Leukemia Treated at Specialized Cancer Centers in California. J Oncol Pract 15 (4): e316-e327, 2019.
Tratamiento de la leucemia linfoblástica aguda infantil recién diagnosticada
Opciones de tratamiento de inducción estándar de la leucemia linfoblástica aguda recién diagnosticada
La opción de tratamiento estándar de la leucemia linfoblástica aguda infantil recién diagnosticada es la siguiente:
- Quimioterapia.
Quimioterapia de inducción a la remisión
El objetivo de la primera fase del tratamiento (inducción a la remisión) es producir una remisión completa (RC). Esta fase de inducción suele durar 4 semanas. En general, cerca del 98 % de los pacientes con diagnóstico reciente de LLA-B alcanzan la RC al final de esta fase; las tasas de remisión son más bajas en los lactantes y en niños mayores con LLA-T, o que tienen recuentos leucocitarios altos durante la presentación inicial.
La quimioterapia de inducción suele incluir los siguientes fármacos, con una antraciclina o sin esta (doxorrubicina o daunorrubicina):
- Vincristina.
- Corticoesteroides (prednisona o dexametasona).
- Asparaginasa.
- Quimioterapia intratecal.
En los protocolos del Children's Oncology Group (COG) se administra una inducción de 3 fármacos (vincristina, un corticoesteroide y pegaspargasa) a los pacientes con LLA-B de riesgo estándar del Instituto Nacional del Cáncer (NCI); y de 4 fármacos (vincristina, un corticoesteroide y pegaspargasa con una antraciclina) a los pacientes con LLA-B de riesgo alto del NCI, así como a todos los pacientes con LLA-T. Otros grupos usan una inducción de 4 fármacos en todos los pacientes.
Terapia con corticoesteroides
En muchos regímenes actuales se usa la dexametasona en lugar de la prednisona durante la inducción a la remisión y en las fases posteriores del tratamiento, aunque hay polémica sobre el beneficio de la dexametasona en todos los subgrupos de pacientes. En algunos ensayos también se indica que la dexametasona durante la inducción tal vez produzca más efectos tóxicos que la prednisona, entre ellos, tasas altas de infecciones, miopatía y cambios de comportamiento. El COG notificó que el uso de dexametasona durante la inducción se relacionó con un riesgo más alto de osteonecrosis en niños de más edad (edad >10 años), aunque este hallazgo no se corroboró en otros estudios aleatorizados.
Evidencia (dexametasona vs. prednisona durante la inducción):
- El Children’s Cancer Group dirigió un ensayo aleatorizado en el que se comparó la dexametasona y la prednisona en pacientes con LLA-B de riesgo estándar que recibieron una inducción de 3 fármacos sin antraciclina.
- La dexametasona se relacionó con una supervivencia sin complicaciones (SSC) superior.
- También se relacionó con una frecuencia más alta de miopatía por corticoesteroides reversible e hiperglucemia. No se observaron diferencias significativas en las tasas de infecciones durante la inducción en los 2 grupos aleatorizados.
- El Medical Research Council (MRC) del Reino Unido dirigió otro ensayo aleatorizado con pacientes de riesgo estándar y de riesgo alto.
- En este ensayo se demostró que la dexametasona se relaciona con un desenlace más favorable que la prednisolona en todos los subgrupos de pacientes.
- La incidencia de recaídas en el sistema nervioso central (SNC) y en otros sitios fue significativamente inferior en los pacientes que recibieron dexametasona que en aquellos que recibieron prednisolona.
- La dexametasona se relacionó con mayor incidencia de problemas de comportamiento relacionados con los corticoesteroides y miopatía, pero no se observó ningún exceso de riesgo de osteonecrosis. No hubo diferencia en las tasas de mortalidad durante la inducción entre los grupos aleatorizados.
- En el ensayo de la Associazione Italiana di Ematologia e Oncologia Pediatrica (AIEOP) ALL-BFM-2000 (NCT00430118) 3720 pacientes se asignaron al azar a recibir dexametasona (10 mg/m2/d) o prednisona (60 mg/m2/d) durante una inducción a la remisión multifarmacológica (que incluyó una antraciclina en todos los pacientes) después de una profase con prednisona de 7 días.
- La dexametasona se relacionó con mayor incidencia de episodios potencialmente mortales (sobre todo de infecciones), lo que se tradujo en una tasa de mortalidad significativamente más alta (2,5 % para la dexametasona vs. 0,9 % para la prednisona; P = 0,00013).
- No hubo diferencia en las tasas de osteonecrosis entre los grupos aleatorizados.
- La incidencia acumulada de recaída a 5 años fue significativamente más baja con la dexametasona (11 vs. 16 %; P< 0,0001), lo que produjo tasas de SSC a 5 años superiores (84 % para la dexametasona vs. 81 % para la prednisona, P = 0,024), a pesar del aumento en la tasa de mortalidad durante la inducción.
- No se observaron diferencias en la supervivencia general (SG) de acuerdo con la aleatorización por tipo de corticoesteroide, aunque el estudio no tuvo una potencia suficiente como para detectar diferencias pequeñas en la SG.
- En un análisis por subgrupos predefinido, se observó un beneficio de supervivencia con el tratamiento de dexametasona en los pacientes con LLA-T y una respuesta favorable a la profase con prednisona (tasas de SG a 5 años, 91 % para dexametasona vs. 83 % para prednisona, P = 0,036).
- El COG dirigió un ensayo aleatorizado de dexametasona y prednisona en pacientes con LLA-B de riesgo alto del NCI. Los pacientes se asignaron al azar a recibir 14 días de dexametasona o 28 días de prednisona durante una inducción de 4 fármacos (incluso una antraciclina). En este ensayo también se incluyó una comparación aleatorizada de metotrexato en dosis altas o en aumento escalonado durante la fase de mantenimiento provisional.
- La dexametasona se relacionó con una tasa más alta de infecciones, pero no hubo diferencia en la tasa de mortalidad durante la inducción entre la dexametasona y la prednisona.
- En los pacientes que tenían menos de 10 años en el momento del diagnóstico, hubo una interacción estadística significativa entre los grupos de asignación al tratamiento con corticoesteroide y con metotrexato. Sin embargo, el mejor resultado dentro de este grupo de pacientes se observó en aquellos que recibieron dexametasona durante la inducción y dosis altas de metotrexato durante el mantenimiento provisional.
- La aleatorización con corticoesteroides se cerró antes de tiempo para pacientes de 10 años o más en el momento del diagnóstico debido a las tasas excesivas de osteonecrosis en pacientes asignados al azar para recibir dexametasona. Sin embargo, no hubo ningún beneficio en la SSC relacionado con la dexametasona en estos pacientes mayores (tasas de SSC a 5 años del 73,1 % con dexametasona y del 73,9 % con prednisona; P = 0,78).
La relación entre las dosis de dexametasona y prednisona quizás afecta el desenlace. En los estudios en los que la relación entre las dosis de dexametasona y prednisona fue de 1:5 a 1:7, se observó un mejor resultado para la dexametasona, mientras que en los estudios en los que se usó una relación de 1:10 se observaron resultados similares.
Asparaginasa
Las siguientes son las formas de asparaginasa que se han usado en el tratamiento de los niños con LLA:
- Pegaspargasa (PEG-asparaginasa).
- Calaspargasa pegol.
- Asparaginasa de Erwinia chrysanthemi (Erwinia L-asparaginasa).
- L-asparaginasa natural de Escherichia coli (E. coli) (no está disponible en los Estados Unidos, pero sí en otros países).
Pegaspargasa (PEG-asparaginasa)
La pegaspargasa, una forma de L-asparaginasa en la que se modifica la enzima derivada de E. coli mediante un enlace covalente de polietilenglicol, se usa con frecuencia durante las fases de inducción y posinducción del tratamiento en pacientes con diagnóstico reciente en Europa occidental. La pegaspargasa no está disponible en los Estados Unidos, pero sí en otros países.
La pegaspargasa se administra por vía intramuscular (IM) o intravenosa (IV). La farmacocinética y los perfiles de toxicidad son similares para la administración IM o IV de la pegaspargasa. No se ha comprobado que la administración IV de la pegaspargasa sea más tóxica que la administración IM.
La pegaspargasa tiene una semivida en suero mucho más larga que la L-asparaginasa natural de E. coli, lo que produce una disminución prolongada de la asparagina después de una sola inyección.
Los índices de actividad enzimática de la asparaginasa sérica superiores a 0,1 UI/ml se relacionan con disminución de la asparagina sérica. En algunos estudios se observó que una sola dosis de pegaspargasa por vía IM o IV, como parte de una inducción multifarmacológica, produce un índice de actividad enzimática en suero superior a 0,1 UI/ml en casi todos los pacientes durante, por lo menos, 2 a 3 semanas. En un estudio de 54 pacientes de riesgo alto del NCI que realizó el COG, una actividad baja de la asparaginasa en plasma hasta de 0,02 UI/ml se relacionó con disminución de la asparagina sérica. Al usar el valor de corte, se calculó que en el 96 % de los pacientes se mantenía el efecto terapéutico (disminución de la asparagina en plasma) durante los 22 a 29 días posteriores a una única dosis de pegaspargasa de 2500 UI/m2. En un estudio aleatorizado, el uso de dosis más altas de pegaspargasa (3500 UI/m2) no mejoró el resultado en comparación con el uso de dosis estándar (2500 UI/m2).[Nivel de evidencia A1]
En otro estudio, se redujeron las dosis de pegaspargasa con el fin de disminuir la toxicidad. Y si bien estas dosis más bajas permitieron mantener índices adecuados de asparaginasa de más de 0,1 UI/ml, la frecuencia de efectos tóxicos relacionados con la asparaginasa fue similar a la frecuencia de efectos tóxicos notificada en estudios previos en los que se usaron dosis de pegaspargasa más elevadas. En este estudio no se informó sobre el efecto de las dosis más bajas de pegaspargasa en la SSC.
Evidencia (uso de la pegaspargasa vs. L-asparaginasa natural de E. coli):
- Se llevó a cabo una comparación aleatorizada de pegaspargasa IV versus asparaginasa natural de E. coli. Cada sustancia se administró durante un periodo de 30 semanas después de alcanzar la RC.[Nivel de evidencia A3]
- El índice de actividad de la asparaginasa sérica (SAA) fue significativamente más alto con la pegaspargasa IV y superó el objetivo terapéutico (>0,1 UI/ml) en casi todos los pacientes durante el periodo de 30 semanas.
- No se presentaron diferencias significativas en la SSC y la SG entre los grupos aleatorizados.
- Tampoco se presentaron diferencias en las tasas de efectos tóxicos relacionados con la asparaginasa, como la hipersensibilidad, la pancreatitis y las complicaciones tromboembólicas.
- En ambos grupos de pacientes se observaron semejanzas en los resultados y las tasas de efectos tóxicos de la asparaginasa.
- Según la evaluación de las encuestas de pacientes y progenitores, la pegaspargasa IV se relacionó con menos ansiedad vinculada con el tratamiento.
- En otro ensayo aleatorizado, los pacientes con LLA de riesgo estándar se asignaron a recibir pegaspargasa o asparaginasa natural de E. coli durante la inducción y en cada uno de los 2 cursos de intensificación diferida.
- La actividad y la toxicidad de una dosis única de pegaspargasa con vincristina y prednisona durante la terapia de inducción fueron similares a 9 dosis IM de L-asparaginasa de E. coli (3 veces por semana durante 3 semanas).
- El uso de pegaspargasa se relacionó con una eliminación más rápida de los blastocitos y una incidencia más baja de anticuerpos neutralizantes.
Los pacientes con reacciones alérgicas a la pegaspargasa se suelen cambiar a L-asparaginasa de Erwinia. En un análisis del COG se investigó el efecto perjudicial de la interrupción prematura del tratamiento con pegaspargasa sobre la supervivencia sin enfermedad (SSE) en pacientes con LLA-B de riesgo alto. En el estudio se observó que el efecto indeseable sobre el desenlace se podía revertir si se usaba la L-asparaginasa de Erwinia para completar el curso de tratamiento con asparaginasa previsto.[Nivel de evidencia C2] Las mediciones de los índices de SAA después de una reacción leve o dudosa a la pegaspargasa quizás permitan diferenciar entre los pacientes que se deben cambiar a la L-asparaginasa de Erwinia (debido a SAA insuficiente) y aquellos que no necesitan el cambio de formulación.
Evidencia (efecto pronóstico adverso de la interrupción prematura de pegaspargasa o de la inactivación asintomática de asparaginasa):
- En varios estudios se identificó a un subgrupo de pacientes que presentaron una inactivación asintomática de la asparaginasa, que se define como la ausencia de índices terapéuticos de SAA sin manifestaciones alérgicas.
- En un ensayo dirigido por el Dana-Farber Cancer Institute (DFCI) Consortium, el 12 % de los pacientes que al inicio recibieron L-asparaginasa natural de E.coli presentaron una inactivación asintomática. Estos pacientes tuvieron una SSC superior cuando se les cambió la formulación de asparaginasa.
- Los pacientes que recibieron pegaspargasa presentaron índices más bajos de inactivación asintomática (<10 %).
Se necesita investigar más la frecuencia óptima para la vigilancia farmacocinética de los pacientes tratados con pegaspargasa, y si esto afecta el desenlace.
- En un informe del COG se incluyeron 8196 pacientes con LLA-B recién diagnosticada que se inscribieron entre 2004 y 2011.[Nivel de evidencia C2]
- La incidencia acumulada de la interrupción de pegaspargasa (debido a toxicidad) fue del 12,2 % en los pacientes de riesgo estándar del NCI y del 25,4 % en los pacientes de riesgo alto del NCI.
- En los análisis multivariantes, los pacientes de riesgo alto del NCI que interrumpieron prematuramente el tratamiento con pegaspargasa tuvieron una SSE inferior (cociente de riesgos instantáneos [CRI], 1,5; P = 0,002) que aquellos que recibieron todas las dosis indicadas. En los pacientes de riesgo estándar del NCI, la interrupción de la pegaspargasa no tuvo ningún efecto sobre la SSE, excepto en los pacientes con una respuesta temprana lenta que recibieron una terapia de posinducción intensificada (CRI, 1,7; P = 0,03).
- Los pacientes de riesgo alto del NCI que interrumpieron el tratamiento con pegaspargasa, pero que luego se cambiaron a asparaginasa de Erwinia y recibieron todas las dosis posteriores indicadas, no tuvieron un mayor riesgo de recaída (CRI, 1,1; P = 0,69).
- En un análisis del protocolo ALL2008 de la Nordic Society of Pediatric Hematology and Oncology (NOPHO), en el que se incluyeron 1115 pacientes con LLA que no presentaban riesgo alto, se notificó lo siguiente:
- De los pacientes, 255 recibieron un curso incompleto de asparaginasa debido a la toxicidad, y 46 pacientes presentaron indicios de inactivación asintomática durante el seguimiento del fármaco terapéutico.
- La incidencia acumulada de recaída a 7 años fue del 11,1 % en los 301 pacientes que recibieron un curso de asparaginasa incompleto, y del 6,7 % en los 814 pacientes restantes que recibieron los cursos terapéuticos previstos (CRI, 1,73; P = 0,03).
- En un modelo de Cox, el tratamiento con asparaginasa subóptimo (debido a un tratamiento con pegaspargasa interrumpido o a inactivación asintomática) se relacionó de forma significativa con un mayor riesgo de recaída (CRI, 1,69; P = 0,03).
En un intento por disminuir las reacciones de hipersensibilidad a la pegaspargasa, en el protocolo Dutch Childhood Oncology Group-ALL11 se asignó al azar a los pacientes para recibir dosis continuas o discontinuas después de la terapia de inducción. En el grupo de dosis continuas, el número de reacciones de hipersensibilidad o inactivación silenciosa fue 7 veces menor y las concentraciones de anticuerpos fueron significativamente inferiores. No hubo diferencias entre los grupos de tratamiento en el número total de efectos tóxicos producidos por la asparaginasa ni en las incidencias a 5 años de recaída, mortalidad y supervivencia sin enfermedad.
Calaspargasa pegol
La calaspargasa pegol es otra formulación de asparaginasa pegilada que también está disponible para el tratamiento de los niños y adolescentes con LLA. Esta formulación tiene una estructura similar a la de la pegaspargasa, excepto por un enlace diferente entre la enzima L-asparaginasa y la fracción PEG, lo que se traduce en una semivida más larga.
Evidencia (calaspargasa pegol vs. pegaspargasa):
- En un estudio del COG, 165 pacientes con LLA-B de riesgo alto se asignaron al azar para recibir calaspargasa pegol o pegaspargasa durante la fase de inducción del tratamiento de la LLA.
- La semivida media de la calaspargasa pegol fue cerca de 2,5 veces más larga que la de la pegaspargasa.
- La exposición sistémica total a la calaspargasa pegol fue mayor que a la pegaspargasa.
- A los 25 días de la dosis con calaspargasa pegol, el 95 % de los pacientes presentaba índices de asparaginasa de más de 0,1 UI/ml , en comparación con el 28 % de los pacientes que recibieron pegaspargasa.
- La evidencia de enfermedad residual mínima (ERM) negativa al final de la inducción fue similar entre los 2 fármacos (74 % y 72 %).
- El perfil de toxicidad de ambos fármacos fue parecido.
- En un ensayo del DFCI de calaspargasa pegol en pacientes con LLA recién diagnosticada, todos los participantes recibieron una dosis de calaspargasa pegol o pegaspargasa como parte de la terapia de inducción. Después de la inducción, 230 pacientes se asignaron al azar para recibir calaspargasa pegol cada 3 semanas (10 dosis) o pegaspargasa cada 2 semanas (15 dosis).
- En el día 25 a partir de la dosis de inducción, el 88 % de los pacientes que recibieron calaspargasa pegol tuvieron un índice de asparaginasa de 0,1 UI/ml, en comparación con el 17 % de los pacientes que recibieron pegaspargasa.
- No hubo diferencia en la ERM al final de la inducción.
- No hubo diferencia en la frecuencia de efectos tóxicos (37 %).
- Las tasas de SSC a 5 años con la calaspargasa pegol y la pegaspargasa fueron similares (88,1 vs. 84,9 %).
En los Estados Unidos, el uso de la calaspargasa pegol está aprobado solo para pacientes menores de 22 años.
Asparaginasa de Erwinia chrysanthemi (L-asparaginasa de Erwinia )
La L-asparaginasa de Erwinia se suele usar en pacientes con alergia a la L-asparaginasa natural de E. coli o a la pegaspargasa.
La semivida de la L-asparaginasa de Erwinia (0,65 días) es mucho más corta que la semivida de la asparaginasa natural de E. coli (1,2 días) o de la pegaspargasa (5,7 días). Si se usa la L-asparaginasa de Erwinia, la semivida más corta de la formulación con Erwinia exige una administración más frecuente para lograr una reducción adecuada de la asparagina.
Evidencia (aumento de la frecuencia de la dosis de Erwinia necesario para lograr el efecto terapéutico previsto):
- En un ensayo del COG se demostró que la administración IM de la L-asparaginasa de Erwinia 3 veces por semana en los pacientes con alergia a la pegaspargasa conduce a índices terapéuticos de actividad enzimática de la asparaginasa sérica (definidos como un índice ≥0,1 UI/ml).
- En ese ensayo, el 96 % de los niños alcanzó un índice de 0,1 UI/ml o más 2 días después de una dosis de L-asparaginasa de Erwinia y el 85 % logró el mismo índice 3 días después la dosis.
- En un ensayo de administración IV de L-asparaginasa de Erwinia (programa lunes-miércoles-viernes) a pacientes con alergia a la pegaspargasa, se encontró un índice terapéutico de actividad enzimática de la asparaginasa sérica (definida como ≥0,1 UI/ml) en el 83 % de los pacientes a las 48 horas de la dosis, pero solo en el 43 % de los pacientes a las 72 horas de la dosis.
- Si la formulación IV de Erwinia se administra en un programa lunes-miércoles-viernes, los autores recomiendan que se vigilen los índices de actividad de la enzima durante 72 horas con el fin de asegurar concentraciones terapéuticas.
En un ensayo de fase II/III del COG se estudió una forma recombinante de L-asparaginasa de Erwinia, la asparaginasa erwinia chrysanthemi (recombinante)-rywn. Cuando se administró un programa lunes (25 mg/m2), miércoles (25 mg/m2) y viernes (50 mg/m2) durante 6 dosis, la proporción de pacientes que alcanzaron una concentración de asparaginasa de 0,1 UI/ml o superior fue del 90 % a las 72 horas (44 de 49 pacientes) y del 96 % a las 48 horas (47 de 49 pacientes). Este perfil de inocuidad fue semejante al de otras formas de asparaginasa. En 2022, la Administración de Alimentos y Medicamentos de los Estados Unidos aprobó la asparaginasa de Erwinia chrysanthemi (recombinante)-rywn para uso IM en niños y adultos con LLA en el programa de lunes, miércoles y viernes que se usó en el ensayo del COG.
Uso de antraciclinas durante la inducción
En los protocolos del COG se administra una inducción de 3 fármacos (vincristina, un corticoesteroide y pegaspargasa) a los pacientes con LLA-B de riesgo estándar del NCI; y de 4 fármacos (vincristina, un corticoesteroide y pegaspargasa con una antraciclina) a los pacientes con LLA-B de riesgo alto del NCI y a todos los pacientes con LLA-T. Otros grupos usan una inducción de 4 fármacos en todos los pacientes.
Por lo general se utiliza daunorrubicina o doxorrubicina para los regímenes de inducción que incluyen una antraciclina. En un ensayo aleatorizado de comparación entre ambos fármacos durante la inducción, no se encontraron diferencias en las mediciones de respuesta temprana, como la disminución del recuento de blastocitos periféricos durante la primera semana de tratamiento, las características morfológicas de la médula el día 15 y los índices de ERM al final de la inducción.[Nivel de evidencia B3]
Respuesta a la quimioterapia de inducción a la remisión
Más del 95 % de los niños con LLA recién diagnosticada alcanzarán una RC durante las primeras 4 semanas de tratamiento. Entre los que no alcanzan la RC en el transcurso de las primeras 4 semanas, cerca de la mitad morirá debido a toxicidad durante la fase de inducción (a menudo, por infecciones) y el resto exhibirá una enfermedad resistente (leucemia persistente en la evaluación morfológica).; [Nivel de evidencia C1]
Tradicionalmente, la remisión se define como un examen de médula ósea al final de la inducción mediante un estudio citomorfológico microscópico de rutina con menos del 5 % de linfoblastos al final de la inducción (M1). El consorcio Ponte de Legno incluye alrededor de 15 grupos cooperativos grandes nacionales e internacionales que se dedican al estudio y tratamiento de la LLA infantil. Este grupo publicó por consenso la siguiente definición para la remisión completa:
- Índices de ERM menores al 1 % y características morfológicas M1.
- La ERM es el criterio de referencia y tiene preferencia frente a las características citomorfológicas.
- La ERM se determina por técnicas de citometría de flujo o de reacción en cadena de la polimerasa.
- Resolución de la enfermedad extramedular, que se evalúa recién al final de la inducción.
La mayoría de los pacientes que presentan una leucemia detectable persistente en las evaluaciones morfológicas al final de la fase de inducción de 4 semanas tiene un pronóstico precario y quizás se beneficie de un trasplante de células madre hematopoyéticas (TCMH) alogénico una vez que se obtenga la RC. En un estudio grande, 1041 pacientes con enfermedad persistente después de la inducción (fracaso de la inducción) tratados entre 1985 y 2000, la tasa de SG a 10 años fue del 32 %. Se observó una tendencia a un desenlace superior para el TCMH alogénico, en comparación con la quimioterapia sola, en los pacientes con un fenotipo de células T (cualquier edad) y en los pacientes mayores de 6 años con LLA-B. Los pacientes con LLA-B de 1 a 5 años de edad en el momento del diagnóstico que carecían de anomalías citogenéticas adversas (reordenamiento de KMT2A, BCR::ABL1) tuvieron un pronóstico relativamente favorable, sin ninguna ventaja para el desenlace cuando recibieron un TCMH en comparación con la quimioterapia sola.
En un estudio retrospectivo de seguimiento se notificaron los desenlaces de 325 niños y adolescentes con LLA-T y fracaso de la inducción inicial tratados entre 2000 y 2018. La tasa de SG a 10 años fue del 54,7 %, significativamente mejor que las tasas de los controles históricos tratados entre 1985 y 2000 (tasa de SG a 10 años, 27,6 %). Con el tiempo, el 93 % de los pacientes con LLA-T y fracaso de la inducción inicial alcanzó la remisión completa. Entre quienes alcanzaron la remisión completa, el 72 % recibió un TCMH. Después de ajustar por el tiempo transcurrido hasta el trasplante, la tasa de SG a 10 años fue del 66,2 % para estos pacientes en comparación con el 50,8 % de los pacientes que no recibieron trasplantes.
La incorporación de nelarabina quizás sea importante para los pacientes de LLA-T con fracaso de la inducción. En el estudio del COG AALL0434 (NCT00408005) se incluyeron 43 pacientes que presentaban más de un 25 % de blastocitos en un aspirado de médula ósea al final de la inducción. De estos pacientes, 23 se asignaron de manera no aleatorizada a una terapia que incluyó dosis altas de metotrexato y nelarabina como parte de un régimen multifarmacológico, y 20 pacientes se sometieron a un trasplante alogénico. La tasa de SSC a 5 años fue del 53,1 % (± 9,4 %) en los pacientes que recibieron dosis altas de metotrexato y nelarabina. No hubo diferencias en el desenlace de los 2 grupos (CRI, 0,66; IC 95 %, 0,24–1,83; P = 0,423).
En los pacientes que alcanzaron una RC, las mediciones de la velocidad de eliminación de los blastocitos y de la ERM tienen gran importancia pronóstica, en especial, en los siguientes casos:
- El porcentaje de blastocitos detectables en la médula ósea mediante estudios morfológicos a los 7 y 14 días del inicio de la terapia multifarmacológica de inducción a la remisión se ha relacionado con el riesgo de recaída, y el COG lo ha usado para estratificar el riesgo de los pacientes. Sin embargo, cuando se incluye la ERM al final de la inducción en los análisis multivariantes, estos hallazgos medulares tempranos pierden su importancia pronóstica.
- El desenlace a largo plazo se correlaciona de forma estrecha con la presencia de índices elevados de ERM submicroscópica al final de la inducción detectados mediante citometría de flujo multiparamétrica, reacción en cadena de la polimerasa o secuenciación de última generación. La intensificación de la terapia de posinducción en los pacientes con índices altos de ERM al final de la inducción es un componente frecuente de la mayoría de los regímenes de tratamiento para la LLA. En un ensayo aleatorizado dirigido por el grupo United Kingdom Acute Lymphoblastic Leukaemia (UKALL), se observó que la terapia de posinducción intensificada mejora el desenlace en los pacientes de riesgo estándar y riesgo intermedio que tienen una ERM alta al final de la inducción.
- Los índices de ERM al comienzo de la inducción (por ejemplo, en los días 8 y 15) y después durante la posinducción (por ejemplo, a las 12 semanas del inicio del tratamiento) también tienen importancia pronóstica para los pacientes con LLA-B y LLA-T.
- Casi todos los pacientes con ERM positiva al final de la inducción tendrán una ERM negativa al final de 4 a 8 semanas de terapia de consolidación. En un estudio del COG, los pacientes con LLA-B de riesgo alto que presentaron una ERM positiva al final de la inducción, pero una ERM negativa al final de la consolidación, tuvieron una SSE significativamente mejor que los pacientes que presentaron una ERM positiva al final de la consolidación (tasa de SSE a 5 años, 79,5 vs. 39,5 %).
Para obtener más información, consultar la sección Respuesta al tratamiento inicial.
Para obtener información específica sobre la terapia dirigida al SNC para prevenir la recaída en el SNC de niños con LLA recién diagnosticada, consultar la sección Terapia dirigida al sistema nervioso central para la leucemia linfoblástica aguda infantil.
Opciones de tratamiento de posinducción estándar de la leucemia linfoblástica aguda infantil
Las opciones de tratamiento estándar para el tratamiento de consolidación o intensificación y mantenimiento (terapia de posinducción) son las siguientes:
- Quimioterapia.
Todos los grupos administran terapia dirigida al SNC durante la quimioterapia premantenimiento. En algunos protocolos (COG, St. Jude Children's Research Hospital [SJCRH] y DFCI), se administra quimioterapia intratecal continua durante el mantenimiento, mientras que en otros (Berlin-Frankfurt-Münster [BFM]) no. Para obtener información específica sobre la terapia dirigida al SNC para prevenir la recaída en el SNC de niños con leucemia linfoblástica aguda (LLA) que están recibiendo terapia de posinducción, consultar la sección Terapia dirigida al sistema nervioso central para la leucemia linfoblástica aguda infantil.
Terapia de consolidación o intensificación
Una vez que se alcanza la RC, se administra tratamiento sistémico junto con una terapia dirigida al SNC. La intensidad de la quimioterapia de posinducción varía mucho según la asignación al grupo de riesgo, pero todos los pacientes reciben alguna forma de intensificación después de lograr la RC y antes de iniciar la terapia de mantenimiento.
El programa de intensificación que más se usa es el tratamiento de base del BFM. El grupo de ensayos clínicos BFM fue el pionero en este tratamiento de base, que incluye los siguientes aspectos:
- Una consolidación inicial (llamada inducción IB) justo después de la fase de inducción inicial. Esta fase incluye terapia intratecal, ciclofosfamida, dosis bajas de citarabina y mercaptopurina.
Una fase de mantenimiento provisional, que incluye terapia intratecal y 4 dosis altas de metotrexato (a menudo, 5 g/m2) con rescate de leucovorina.
- Reinducción (o intensificación diferida), que suele incluir fármacos y programas similares a los utilizados durante las fases de inducción y de consolidación inicial.
- Mantenimiento, que suele incluir mercaptopurina (6-MP) diaria, dosis bajas semanales de metotrexato y, a veces, la administración intermitente de vincristina y un corticoesteroide, así como terapia intratecal continuada.
Muchos grupos han adoptado este tratamiento de base, incluso el COG. Las siguientes son las variaciones de este tratamiento de base:
- Intensificación para los pacientes de riesgo más alto con dosis adicionales de vincristina y pegaspargasa, así como la repetición de las fases de mantenimiento provisional y de intensificación diferida.
- El uso de dosis escalonadas de metotrexato (dosis iniciales de 100 mg/m2) sin rescate de leucovorina en lugar de dosis altas de metotrexato, o además de estas dosis, durante las fases de mantenimiento provisional.
- Eliminación o acortamiento de algunas de las fases para los pacientes de riesgo más bajo con el fin de reducir al mínimo la toxicidad aguda y a largo plazo.
Otros grupos de ensayos clínicos utilizan un tratamiento de base diferente durante las fases de posinducción:
- DFCI. Los protocolos del DFCI ALL Consortium incluyen la administración de pegaspargasa durante 30 semanas desde la semana 7 del tratamiento, y un régimen de mantenimiento (dosis altas repetidas de vincristina y dexametasona, dosis bajas de metotrexato semanales y mercaptopurina diaria). Estos protocolos tampoco incluyen una fase de intensificación diferida, pero los pacientes de riesgo alto reciben dosis adicionales de doxorrubicina (en lugar de dosis bajas de metotrexato) durante los primeros 6 meses de la terapia de posinducción.
- NOPHO. El NOPHO también destaca el uso de pegaspargasa durante la consolidación y la intensificación. En el ensayo ALL2008, todos los pacientes recibieron 5 dosis de pegaspargasa en semanas alternas después de la inducción. Luego, los pacientes recibieron 10 dosis adicionales cada 2 semanas o 3 dosis cada 6 semanas. Ambos regímenes produjeron tasas excelentes de supervivencia similares; la toxicidad fue menor en el régimen de 3 dosis.
- SJCRH. En el SJCRH se sigue el tratamiento de base del BFM, pero se intensifican las fases de reinducción y mantenimiento para algunos pacientes al incluir una dosificación intensificada de pegaspargasa, dosis altas repetidas frecuentes de vincristina y corticoesteroides, y rotación de díadas de fármacos durante el mantenimiento (mercaptopurina y metotrexato, ciclofosfamida y citarabina, dexametasona y vincristina).
Leucemia linfoblástica aguda de riesgo estándar
En los niños con LLA de riesgo bajo y de riesgo estándar se ha intentado limitar la exposición a fármacos, como las antraciclinas y los alquilantes, que se relacionan con un aumento del riesgo de efectos tóxicos tardíos. El régimen COG para la terapia de posinducción de la LLA-B de riesgo estándar se puede administrar en un entorno ambulatorio y tiene varias características favorables, como la consolidación de 4 semanas de baja intensidad, reducción de la exposición a antraciclinas (75 mg/m2) y a alquilantes (1 g/m2), solo 2 dosis de pegaspargasa, y fases de mantenimiento provisional que consisten en dosis escalonadas de metotrexato (sin rescate de leucovorina) en lugar de dosis altas de metotrexato IV.[Nivel de evidencia B4]
También se notificaron resultados favorables para los pacientes con LLA-B de riesgo estándar en ensayos en los que se usó un número limitado de cursos de metotrexato en dosis intermedias o altas durante la consolidación seguida de terapia de mantenimiento (sin fase de reinducción). En concreto, un subgrupo de pacientes con LLA-B de riesgo estándar con características citogenéticas favorables, sin indicios de enfermedad testicular o del SNC en el momento del diagnóstico y que alcanzaron pronto índices bajos de ERM, han sido tratados con exposición a dosis bajas o nulas de antraciclinas y alquilantes. La tasa de SSE a 5 años fue casi del 99 %, y la tasa de SG fue del 100 %. Además, en el estudio DFCI ALL Consortium se usaron dosis múltiples de pegaspargasa (30 semanas) como consolidación, sin exposición a alquilantes ni antraciclinas durante la fase de posinducción.
Sin embargo, el efecto pronóstico de la ERM al final de la inducción o la consolidación afectó el tratamiento de los pacientes asignados en un comienzo al grupo de riesgo estándar del NCI. En varios estudios se demostró que los índices más altos de ERM al final de la inducción se relacionan con un pronóstico más precario. Se ha observado que la intensificación del tratamiento mejora el desenlace en los pacientes de riesgo estándar con un índice de ERM alto al final de la inducción. A los pacientes con LLA-B de riesgo estándar del NCI con características de riesgo alto (como el aumento de los índices de ERM al final de la inducción, así como el estado SNC2 en el momento del diagnóstico y las características genéticas desfavorable) se les administra una terapia más intensiva. Para obtener más información, consultar la sección Grupos pronósticos (riesgo) en evaluación clínica.
Evidencia (intensificación para la LLA-B de riesgo estándar):
- En ensayos clínicos de la década de 1980 y principios de la década de 1990, se observó que el uso de una fase de intensificación diferida mejoró el desenlace de los niños con LLA de riesgo estándar tratados con regímenes que incluyen el tratamiento de base del BFM. La fase de intensificación diferida de este tipo de regímenes, como los del COG, consiste en una fase de reinducción de 8 semanas (que incluye dexametasona y una antraciclina), así como una fase de reconsolidación con ciclofosfamida, citarabina y 6-tioguanina administradas 4 a 6 meses después de alcanzar la remisión.
- En el antiguo estudio del Children's Cancer Group (CCG) (CCG-1991/COG-1991) de LLA de riesgo estándar, se usó dexametasona en una fase de inducción de 3 fármacos y se probó la utilidad de una segunda fase de intensificación diferida. En este estudio también se comparó el aumento escalonado de las dosis IV de metotrexato (sin rescate de leucovorina) con vincristina versus una combinación de mantenimiento estándar de metotrexato oral durante 2 fases de mantenimiento provisional.[Nivel de evidencia B1]
- Una segunda fase de intensificación diferida no ofreció ningún beneficio a los pacientes que presentaron una respuesta temprana rápida (médula de tipo M1 o M2 en el día 14 de la inducción).
- El aumento escalonado del metotrexato IV durante las fases de mantenimiento provisional, en comparación con el metotrexato oral, produjo un aumento significativo de la SSC debido a disminución en la incidencia de recaídas extramedulares aisladas, en especial, las que comprometen el SNC.
- Los tratamientos exitosos para pacientes con LLA de riesgo estándar en los que se redujo el uso de medicamentos relacionados con efectos tóxicos a largo plazo se centraron en niños con LLA-B, no con LLA-T. El COG, el Dutch Children's Oncology Group (DCOG), el DFCI, el NOPHO y otros grandes grupos cooperativos han excluido a los pacientes con LLA-T de los tratamientos de riesgo estándar y riesgo bajo. Los pacientes con características de riesgo estándar del NCI, pero con un inmunofenotipo de células T, presentaron SSC y SG inferiores en comparación con los pacientes con LLA-B de riesgo estándar del NCI tratados con los mismos regímenes en el CCG1952 y el CCG1991.
- En el ensayo del COG AALL0331 (NCT00103285) se estratificó la intensidad del tratamiento para los pacientes de riesgo estándar del NCI a partir de las características biológicas y la respuesta temprana. La respuesta temprana rápida se definió como menos del 5 % de blastocitos en la médula ósea en el día 15 sobre la base de la interpretación morfológica local y una médula de tipo M1 con índices de ERM inferiores al 0,1 % en el día 29. Los pacientes de riesgo estándar bajo fueron los que tenían características biológicas favorables (ETV6::RUNX1 o hiperdiploidía alta con trisomía triple), estado SNC1 y una respuesta temprana rápida. Los pacientes de riesgo estándar promedio fueron los que carecían de características biológicas favorables o desfavorables y que también tuvieron una respuesta temprana rápida. Los pacientes de riesgo estándar alto fueron los que tuvieron una respuesta temprana lenta y estado del SNC3 o pacientes con reordenamiento de KMT2A y respuesta temprana rápida. Todos los pacientes recibieron una inducción de 3 fármacos a base de prednisona (sin antraciclinas). Los pacientes de riesgo estándar promedio fueron asignados al azar a consolidación intensificada (BFM intensificado) o consolidación estándar. Los pacientes de riesgo estándar alto se asignaron en forma no aleatorizada al tratamiento del BFM intensificado completo que se usa para pacientes de riesgo alto del NCI, con 2 fases de intensificación diferida.
- En todos los pacientes, la tasa de SSC a 6 años fue del 89 % y la tasa de SG fue del 96 %.
- Para los pacientes de riesgo estándar bajo, en este estudio se evaluó la incorporación de 4 dosis de pegaspargasa (añadidas en las fases de consolidación y de mantenimiento provisional) al tratamiento estándar, que incluía 2 dosis de pegaspargasa (administradas en las fases de inducción y de intensificación diferida). Los pacientes de riesgo estándar bajo tuvieron resultados muy favorables (las tasas de SSE y de SG a 6 años fueron del 94,7 % ± 0,6 % y del 98,7 % ± 0,3 %, respectivamente). La potenciación de la terapia de riesgo estándar bajo con pegaspargasa adicional no mejoró los desenlaces.[Nivel de evidencia B4]
- Para los pacientes de riesgo estándar promedio, el régimen de consolidación intensificado no mejoró las tasas de remisión completa continua (RCC) o SG. Las tasas a 6 años de RCC y SG para la cohorte de riesgo estándar promedio oscilaron del 88 % al 89 %, y del 95 % al 96 %, respectivamente.
- Los pacientes de riesgo estándar promedio con índices de ERM al final de la inducción del 0,01 a <0,1 % tuvieron un desenlace más precario que aquellos con índices de ERM del <0,01 % (tasas de RCC a 6 años, 77 vs. 91 %, respectivamente). La consolidación intensificada no se asoció con un desenlace superior en pacientes de riesgo estándar promedio con niveles más altos de ERM.
- La cohorte con riesgo estándar alto logró una tasa de RCC a 6 años relativamente favorable del 86 % y una tasa de SG del 93 %.
- En un estudio aleatorizado realizado en el Reino Unido, los niños y adultos jóvenes con LLA sin características de riesgo alto (características citogenéticas adversas o médula de tipo morfológico M3 el día 8 o el día 15 de la inducción) se estratificaron según el riesgo a partir del índice de ERM al final de la inducción (semana 4) y a la semana 11 del tratamiento. Los pacientes con ERM indetectable en la semana 4 (o ERM baja en la semana 4 e indetectable en la semana 11) se consideraron de riesgo bajo y fueron aptos para la asignación al azar a recibir 1 o 2 fases de intensificación diferida.[Nivel de evidencia B1]
- No hubo ninguna diferencia significativa en la SSC de los pacientes que recibieron 1 o 2 fases de intensificación diferida.
- No hubo ninguna diferencia significativa en las muertes relacionadas con el tratamiento entre los 2 grupos; sin embargo, durante la segunda fase de intensificación diferida se presentaron efectos tóxicos de grado 3 o 4 en el 17 % de los 261 pacientes asignados al azar a ese grupo, y un paciente murió por causas relacionadas con el tratamiento durante esa fase.
- En el estudio de la AIEOP ALL-BFM-2000 (NCT00430118) los pacientes de riesgo estándar (definidos como los pacientes con ERM indetectable en los días 33 y 78, sin características citogenéticas de riesgo alto) se asignaron al azar a tratamiento con una fase de intensificación diferida única de intensidad estándar o de intensidad reducida (duración más corta y reducción de las dosis totales de dexametasona, vincristina, doxorrubicina y ciclofosfamida).
- La intensificación diferida de intensidad reducida se relacionó con una tasa de SSE a 8 años inferior (89 vs. 92 %, P = 0,04), producto de un aumento del riesgo de recaída.
- En un análisis de subgrupos que se hizo a pacientes con la fusión ETV6::RUNX1, no se observaron diferencias en el desenlace entre los 2 grupos de tratamiento (tasa de SSE a 8 años, alrededor del 94 % en ambos grupos).
- En el ensayo Malaysia-Singapore ALL MS2010 de pacientes con LLA-B de riesgo bajo, se evaluó un régimen BFM modificado y de menor intensidad. En este caso se omitió la administración de antraciclinas, y se administraron menos dosis del metotrexato de dosis altas y menos dosis de la citarabina de dosis bajas.
- En este ensayo, la SSP a largo plazo (tasa de SSP a 6 años, 96,5 %) no fue inferior en comparación con el resultado del ensayo anterior dirigido por el mismo grupo.
- Este régimen también fue menos tóxico, y produjo una reducción significativa en las tasas de bacteriemia y choque séptico o en la necesidad de admisiones a la unidad de cuidados intensivos.
- Los pacientes que se clasifican como de riesgo intermedio o estándar en el momento del diagnóstico, pero tienen índices altos de ERM al final de la inducción, presentan un pronóstico más precario y se los debe tratar como pacientes de riesgo alto. En el estudio UKALL2003 (NCT00222612) se usó una terapia de posinducción intensificada (dosis adicionales de pegaspargasa y vincristina, y dosis escalonadas de metotrexato IV sin rescate de leucovorina) para el tratamiento de los pacientes de riesgo estándar o riesgo intermedio con índices altos de ERM al final de la inducción.[Nivel de evidencia B1]
- La terapia de posinducción intensificada produjo un aumento de la SSC comparable al de los pacientes con índices bajos de ERM al final de la inducción.
Leucemia linfoblástica aguda de riesgo alto
En los pacientes de riesgo alto, se han usado varios abordajes diferentes con eficacia comparable.; [Nivel de evidencia B4] Por lo general, el tratamiento para los pacientes de riesgo alto es más intensivo que para los pacientes de riesgo estándar y suele incluir dosis acumuladas más altas de varios fármacos, como antraciclinas y alquilantes. El uso de dosis más altas de estos fármacos aumenta el riesgo de toxicidad a corto y largo plazo, y muchos ensayos clínicos se han enfocado en la reducción de los efectos secundarios de estos regímenes intensificados.
Evidencia (intensificación para la LLA de riesgo alto):
- El antiguo CCG formuló una régimen intensificado de tratamiento del BFM que incluyó una segunda fase de mantenimiento provisional y una fase de intensificación diferida. En este régimen se administraban cursos repetidos de dosis escalonadas de metotrexato IV (sin rescate de leucovorina) con vincristina y pegaspargasa durante el mantenimiento provisional, y dosis altas repetidas adicionales de vincristina y pegaspargasa durante la consolidación inicial y la intensificación diferida. En el ensayo CCG-1882, los pacientes de riesgo alto del NCI con respuesta temprana lenta (médula de tipo M3 el día 7 de la inducción) se asignaron al azar a recibir el tratamiento del BFM estándar o intensificado.
- El régimen de tratamiento intensificado del ensayo CCG-1882 produjo tasas de SSC y SG significativamente mejores (75 % y 78 %) que las que se obtienen con el tratamiento del BFM modificado que el CCG usa de manera estándar (55 % y 66,7 %).
- La incidencia de osteonecrosis fue significativamente más alta en los pacientes mayores de 10 años que recibieron el tratamiento intensificado (que incluyó 2 cursos de dexametasona de 21 días durante la posinducción), en comparación con los pacientes del grupo de tratamiento estándar (un curso de dexametasona de 12 días durante la posinducción).
- En un estudio italiano, los investigadores observaron que dos aplicaciones de la terapia de intensificación diferida (protocolo II) mejoraron de manera significativa el desenlace en los pacientes con una respuesta precaria a la profase de prednisona.
- En el ensayo clínico CCG-1961 se usó un diseño factorial de 2 × 2 para comparar el tratamiento estándar, el tratamiento intensificado y el tratamiento de duración estándar (un mantenimiento provisional y una fase de intensificación diferida) con los tratamientos de duración prolongada (2 fases de mantenimiento provisional y de intensificación diferida) en los pacientes de riesgo alto del NCI que exhibieron una respuesta temprana rápida. En este ensayo también se evaluó si la administración continua de dexametasona versus la administración en semanas alternas durante las fases de intensificación diferida afectaba las tasas de osteonecrosis.
- El tratamiento intensificado se relacionó con una mejora de la SSC. No hubo beneficios en la SSC en relación con la administración de la segunda fase de mantenimiento provisional y la fase de intensificación diferida.[Nivel de evidencia A1]
- La incidencia acumulada de osteonecrosis a los 5 años fue del 9,9 % en los pacientes de 10 a 15 años, y del 20,0 % en los pacientes de 16 a 21 años, en comparación con el 1,0 % en los pacientes de 1 a 9 años (P = 0,0001). En los pacientes de 10 a 21 años, la dosificación de dexametasona en semanas alternas durante las fases de intensificación diferida, se relacionó con una incidencia acumulada de osteonecrosis significativamente inferior a la que se obtiene con la dosificación continua (8,7 vs. 17,0 %, P = 0,0005).[Nivel de evidencia A3]
- En el ensayo UKALL2003 (NCT00222612), los pacientes con ERM alta al final de la inducción (>0,01 %) o características citogenéticas de riesgo alto se asignaron al azar a recibir quimioterapia de base BFM de intensidad estándar o intensificada.
- La tasa de SSP a 10 años fue del 87,1 % para los pacientes asignados a quimioterapia de base intensificada en comparación con el 82,1 % para los pacientes asignados a quimioterapia de base de intensidad estándar (P = 0,09).
- Los pacientes con características citogenéticas de riesgo alto presentaron un riesgo significativamente más bajo de recaída cuando recibieron terapia intensificada (tasa de recaída a 10 años, del 22,1 % vs. 52,4 % con la terapia de intensidad estándar; P = 0,016).
- En el estudio del COG AALL0232 (NCT00075725) (2004–2011), los pacientes con LLA-B de riesgo alto recibieron tratamiento de base del BFM intensificado con una fase de mantenimiento provisional y de intensificación diferida. Solo los pacientes con ERM al final de la inducción mayor al 0,1 % o médula de tipo M2 o M3 en el día 15 recibieron 2 fases de mantenimiento provisional o intensificación diferida. Los pacientes se asignaron al azar a recibir dosis altas de metotrexato o dosis escalonadas de metotrexato IV (metotrexato de tipo Capizzi) y pegaspargasa durante la fase de mantenimiento provisional (solo en la primera fase en aquellos que recibieron 2 de estas fases).
- La aleatorización al grupo de metotrexato se interrumpió temprano cuando durante el control provisional previsto se identificó que las dosis altas de metotrexato se relacionaban con un desenlace superior. La SSC a 5 años en los pacientes asignados al azar a dosis altas de metotrexato fue del 79,6 %, en comparación con el 75 % en los pacientes que se asignaron al azar al grupo de metotrexato de tipo Capizzi. Las dosis altas de metotrexato también se relacionaron con una SG a 5 años superior (P = 0,025).
- Los pacientes con ERM inferior al 0,01 % al final de la inducción tuvieron una SSC a 5 años del 87 %, en comparación con el 74 % en aquellos que presentaron una ERM del 0,01 % al 0,1 %. Aquellos con índices de ERM superiores al 0,1 % presentaron una evolución más desfavorable.
- El uso de dosis altas de metotrexato se relacionó con una tasa de SSC superior en pacientes con ERM al final de la inducción superior al 0,01 % (dosis altas de metotrexato, 68 %; metotrexato de tipo Capizzi, 58 %; P = 0,008).
Debido a que el tratamiento de la LLA de riesgo alto es más intensivo, produce un riesgo más alto de efectos tóxicos agudos y a largo plazo, de manera que en varios ensayos clínicos se han evaluado intervenciones para prevenir los efectos secundarios sin que ello afecte de manera adversa la SSC. Las intervenciones en investigación incluyen el uso del cardioprotector dexrazoxano para prevenir la cardiotoxicidad de las antraciclinas y un programa diferente de dosificación de corticoesteroides para reducir el riesgo de osteonecrosis.
Evidencia (efecto cardioprotector del dexrazoxano):
- En un ensayo del DFCI ALL Consortium, los niños con LLA de riesgo alto se asignaron al azar a recibir doxorrubicina sola (30 mg/m2/dosis hasta alcanzar una dosis acumulada de 300 mg/m2) o con dexrazoxano durante las fases de inducción e intensificación de la quimioterapia multifarmacológica.
- El uso del cardioprotector dexrazoxano antes de la doxorrubicina, en comparación con el uso de doxorrubicina sola, produjo mejorías de la fracción de acortamiento ventricular izquierda y de los puntajes Z de la dimensión telesistólica sin efectos adversos en la SSC ni aumento del riesgo de una segunda neoplasia maligna a los 5 años del tratamiento.
- Se observó un efecto protector a largo plazo más alto en las niñas que en los niños.
- En el ensayo POG-9404 los pacientes con LLA-T se asignaron al azar a recibir dexrazoxano o no recibir este cardioprotector antes de cada dosis de doxorrubicina (dosis acumulada de 360 mg/m2).
- No hubo diferencia en la SSC entre los pacientes con LLA-T que recibieron dexrazoxano y los pacientes que no recibieron dexrazoxano (dosis acumulada de doxorrubicina, 360 mg/m2).
- Después de 3 años del diagnóstico inicial, la fracción de acortamiento ventricular izquierda y el grosor de la pared ventricular izquierda fueron significativamente más precarios en los pacientes que recibieron doxorrubicina sola que en los pacientes que recibieron dexrazoxano, lo que indica que el dexrazoxano tuvo un efecto cardioprotector. La frecuencia de los efectos tóxicos de grado 3 y 4 durante el tratamiento fue similar entre los grupos aleatorizados, y no hubo diferencias en la incidencia acumulada de segundas neoplasias malignas.
Evidencia (reducción del riesgo de osteonecrosis):
- En el estudio CCG-1961, se analizó el uso de dosis de dexametasona en semanas alternas durante la intensificación diferida con el objetivo de reducir la frecuencia de la osteonecrosis. Los pacientes con LLA-B de riesgo alto y respuesta morfológica temprana rápida a la terapia de inducción se asignaron al azar a recibir 1 o 2 fases de intensificación diferida. Los pacientes asignados al azar a una fase de intensificación diferida recibieron dosis diarias de dexametasona (21 días consecutivos), mientras que los asignados al azar a 2 fases de intensificación diferida recibieron dosis de dexametasona en semanas alternas (días 0–6 y 14–21) durante cada fase de intensificación diferida.
- Entre los pacientes de 10 años de edad o más en el momento del diagnóstico, aquellos que recibieron 2 fases de intensificación diferida (dosificación de dexametasona en semanas alternas) presentaron un riesgo significativamente inferior de osteonecrosis sintomática (incidencia acumulada a 5 años del 8,7 %, en comparación con el 17 % en los pacientes que recibieron una fase de intensificación diferida con dosificación continua de dexametasona; P = 0,001).
- El efecto más intenso se observó en las mujeres de 16 a 21 años, que exhibieron la incidencia más alta de osteonecrosis con la terapia estándar de dexametasona continua; la incidencia de osteonecrosis en el grupo de dexametasona en semanas alternas fue del 5,6 %, en comparación con el 57,6 % en el otro grupo.
Para obtener más información, consultar la sección Osteonecrosis.
Leucemia linfoblástica aguda de riesgo muy alto
Cerca del 10 % al 20 % de los pacientes con LLA se clasifican como de riesgo muy alto, entre ellos los siguientes:
- Lactantes menores de 1 año, en especial si tienen un reordenamiento del gen KMT2A. Para obtener más información sobre los lactantes con LLA, consultar la sección Lactantes con leucemia linfoblástica aguda.
- Pacientes con anomalías citogenéticas adversas, como BCR::ABL1, TCF3::HLF, reordenamientos del gen KMT2A e hipodiploidía baja (<44 cromosomas).
- Pacientes que alcanzaron la RC, pero que tienen una respuesta temprana lenta al tratamiento inicial, como aquellos con un recuento absoluto de blastocitos elevado después de la profase con corticoesteroides de 7días y aquellos con índices altos de ERM al final de la inducción (semana 4) o en momentos posteriores (por ejemplo, semana 12).
- Pacientes con enfermedad persistente en los estudios morfológicos después de las primeras 4 semanas de tratamiento (fracaso de la inducción), incluso si luego alcanzan una RC.
Pacientes con características de riesgo muy alto que recibieron varios ciclos de quimioterapia intensiva durante la fase de consolidación (por lo general, añadida a las fases de intensificación usuales del tratamiento de base del BFM). Estos ciclos adicionales a menudo incluyen fármacos que no se suelen usar en los regímenes de primera línea para la LLA en pacientes de riesgo estándar y alto, así como dosis altas de citarabina, ifosfamida y etopósido. Sin embargo, incluso con este abordaje intensificado, las tasas de SSC a largo plazo notificadas oscilan entre el 30 % y el 50 % en algunos de estos subconjuntos de riesgo muy alto. El DCOG notificó los desenlaces de 107 pacientes con características de riesgo muy alto que se trataron con 3 a 6 bloques de quimioterapia intensiva en 2 ensayos consecutivos. De estos pacientes, 60 recibieron un TCMH alogénico en la primera RC. La tasa de SSC a 5 años fue del 73 % y la tasa de SG fue del 79 % en todos los pacientes. Con este abordaje de tratamiento intensificado, la incidencia acumulada de mortalidad relacionada con el tratamiento fue del 12,3 %, similar a la incidencia acumulada de recaída, que fue del 13 %.
En ciertos ensayos clínicos, los pacientes de riesgo muy alto también se consideran aptos para recibir un TCMH alogénico durante la primera RC. Sin embargo, hay pocos datos sobre el desenlace de los pacientes de riesgo muy alto tratados con TCMH alogénico durante la primera RC. Hay polémica sobre qué subpoblaciones se podrían beneficiar de un TCMH.
Evidencia (TCMH alogénico durante la primera remisión en pacientes de riesgo muy alto):
- En un estudio de un grupo cooperativo europeo realizado entre 1995 y 2000, los pacientes de riesgo muy alto se clasificaron en una de las siguientes categorías: con enfermedad persistente en estudios morfológicos después de una inducción con 4 fármacos, con fusiones BCR::ABL1 o KMT2A::AFF1, o con respuesta precaria a la profase de prednisona en pacientes con fenotipo de células T o recuentos de glóbulos blancos [GB] >100 000/μl. Estos pacientes se asignaron a recibir un TCMH alogénico durante la primera RC (según la disponibilidad de un donante emparentado con compatibilidad de antígeno linfocitario humano) o quimioterapia intensiva.
- Al usar un análisis por intención de tratar, los pacientes que se asignaron al TCMH alogénico (según la disponibilidad de donante) tuvieron una tasa de SSC a 5 años superior a la de los pacientes asignados a quimioterapia intensiva (57 % ± 7 % con trasplante vs. 41 % ± 3 % con quimioterapia, P = 0,02).
- No hubo diferencias significativas en las tasas de SG (56 % ± 6 % con trasplante vs. 50 % ± 3 % con quimioterapia, P = 0,12).
- Para los pacientes con LLA-T y respuesta precaria a la profase de prednisona, las tasas de SSE y SG fueron significativamente mejores con un TCMH alogénico.
- En una serie retrospectiva numerosa de pacientes con fracaso de la inducción inicial, la tasa de SG a 10 años para los pacientes con leucemia persistente fue del 32 %.
- Se observó una tendencia a un resultado superior con el TCMH alogénico, en comparación con la quimioterapia sola en los pacientes con fenotipo de células T (de todas las edades) y con LLA-B en aquellos mayores de 6 años de edad.
- Los pacientes con LLA-B que tenían de 1 a 5 años de edad en el momento del diagnóstico y no presentaban anomalías citogenéticas adversas (reordenamientos de KMT2A, BCR::ABL1) tuvieron un pronóstico relativamente favorable, sin ventajas en el desenlace cuando se sometían a TCMH, en comparación con la quimioterapia sola.
- En el estudio de la AIEOP ALL-BFM-2000 (NCT00430118) (2000–2006), se clasificó a los pacientes como de riesgo alto si cumplían cualquiera de los siguientes criterios: respuesta precaria a la profase de prednisona, incapacidad de lograr una RC al final del primer mes de tratamiento, índices altos de ERM después de la inducción IB (día 78 del tratamiento) y fusión KMT2A::AFF1. Estos pacientes se asignaron a un TCMH alogénico durante la primera RC según el protocolo, en función de la disponibilidad de donantes y la preferencia del investigador.[Nivel de evidencia B4]
- La tasa de SSC a 5 años de los pacientes que cumplían con los criterios de riesgo alto fue del 58,9 %.
- La tasa de SSC a 5 años fue del 74 % en los pacientes cuya única característica de riesgo alto fue una respuesta precaria a la prednisona. No hubo ninguna diferencia significativa en la SSE (P = 0,31) o la SG (P = 0,91) cuando se comparó el TCMH y la quimioterapia en pacientes con respuesta precaria a la prednisona a quienes se les permitió recibir un TCMH de acuerdo con el protocolo (LLA-T o GB ≥100 000/mm3).
- El resto de los pacientes de riesgo alto (es decir, aquellos con fracaso de la inducción inicial, ERM alta en el día 78 o fusión KMT2A::AFF1) tuvieron tasas de SSC de menos del 50 %. Para estos pacientes no hubo ninguna diferencia estadísticamente significativa en la SSE entre aquellos sometidos a un TCMH (n = 66) y quienes recibieron quimioterapia sola (n = 88), después del ajuste por el tiempo de espera del TCMH (5,7 meses).
- En el protocolo del NOPHO ALL2008 (NCT00819351), los pacientes se sometieron a un TCMH en la primera RC cuando presentaron índices de ERM del 5 % o superiores al final de la inducción, o índices de ERM del 0,1 % o superiores al final de la consolidación. Todos los pacientes que se sometieron a TCMH recibieron al menos 3 bloques de quimioterapia intensiva antes del TCMH para reducir los índices de ERM.
- En el análisis por intención de tratar de 69 pacientes que cumplieron con los criterios para TCMH (10 de los cuales no se sometieron a TCMH), la tasa de SSE a 5 años fue del 78 %.
- Al comparar los pacientes de esta cohorte que recibieron un TCMH y aquellos que no, la recepción de un TCMH no se asoció significativamente con la supervivencia (CRI, 1,4; P = 0,69).
- Entre los pacientes que se sometieron a un TCMH, se observaron desenlaces superiores (mejor SSE y menor incidencia acumulada de recaída) en aquellos que tenían ERM indetectable antes del TCMH.
- En los ensayos ALL-10 y ALL-11 del DCOG, los pacientes con características de riesgo muy alto recibieron un régimen de tratamiento intensificado que incluyó 3 bloques de quimioterapia de dosis alta tras la consolidación. Después de los 3 bloques, 60 pacientes recibieron un TCMH alogénico, y 22 pacientes continuaron con un abordaje de quimioterapia sola, que incluyó otros 3 bloques de dosis altas. En estos ensayos, la enfermedad de riesgo muy alto estuvo determinada por la presencia de cualquiera de las siguientes características: enfermedad morfológica detectable al final de la inducción, EMR alta al final de la consolidación (punto de referencia 2), t(4;11), o respuesta precaria a la profase con prednisona.
- La tasa de SSC a 5 años fue del 72,8 % en todos los pacientes.
- En un análisis de referencia de SSC desde el final del tercer bloque de quimioterapia de dosis altas, no se observaron diferencias en los desenlaces de los pacientes que recibieron TCMH, en comparación con aquellos que solo recibieron quimioterapia.
- En 2 análisis retrospectivos, se investigó la función del TCMH durante la primera RC en pacientes con LLA hipodiploide. En los estudios no se comprobó de manera inequívoca que el TCMH mejorara los desenlaces en los siguientes casos: 1) trasplante en todos los pacientes de LLA hipodiploide, 2) trasplante en los pacientes hipodiploides que se consideran de riesgo alto por tener una ERM alta después de la inducción. En estos estudios no se evaluó la estrategia de realizar un TCMH en pacientes con ERM persistente después de la consolidación, ni tampoco se midió la ERM en el momento del trasplante.
- En un estudio de 306 pacientes con hipodiploidía de 16 grupos cooperativos de LLA tratados entre 1997 y 2013, se analizó un subgrupo de 228 pacientes (42 se sometieron a TCMH) con 44 o menos cromosomas que lograron una remisión.[Nivel de evidencia C2]
- Los factores de pronóstico favorable incluyeron un número de 44 cromosomas (comparado con 43 cromosomas o menos), ERM inferior al 0,01 % después de la inducción y tratamiento en el marco de un protocolo estratificado a partir de la ERM según el que se intensificaba la terapia en pacientes con ERM más alta después de la inducción.
- Después del ajuste por la mediana de tiempo hasta el trasplante, los pacientes con ERM baja que se sometieron a TCMH tuvieron una tasa de SSE del 73,6 %, en comparación con una tasa de SSE del 70 % en aquellos que recibieron quimioterapia sola (P = 0,81). Los pacientes con ERM más alta después de la inducción que se sometieron a TCMH tuvieron una tasa de SSE del 55,9 %, en comparación con una tasa de SSE del 40,3 % en aquellos que se trataron con quimioterapia (P = 0,29).
- El COG publicó un análisis de 113 pacientes evaluables de LLA con hipodiploidía tratados entre 2003 y 2011, entre ellos 61 pacientes que se sometieron a un TCMH durante la primera RC.[Nivel de evidencia C1]
- La tasa de SSC a 5 años fue del 57,4 % en los pacientes sometidos a TCMH y del 47,8 % en los pacientes de las cohortes de quimioterapia (P = 0,49). La tasa de SG fue del 66,2 % en los pacientes sometidos a TCMH y del 53,8 % en los pacientes de las cohortes de quimioterapia (P = 0,34).
- Los pacientes con una ERM alta después de la inducción (≥0,01 %) tuvieron una tasa de SSC a 5 años muy precaria del 26,7 %, sin diferencia entre los pacientes que recibieron un TCMH y quienes recibieron quimioterapia.
- En un estudio de 306 pacientes con hipodiploidía de 16 grupos cooperativos de LLA tratados entre 1997 y 2013, se analizó un subgrupo de 228 pacientes (42 se sometieron a TCMH) con 44 o menos cromosomas que lograron una remisión.[Nivel de evidencia C2]
Terapia de mantenimiento
Terapia de mantenimiento de base
En la mayoría de protocolos, la terapia de mantenimiento de base incluye mercaptopurina oral diaria y metotrexato oral o parenteral semanal. En muchos protocolos, la quimioterapia intratecal para el santuario en el SNC continúa durante la terapia de mantenimiento. Además, algunos grupos usan vincristina y corticoesteroides en dosis altas repetidas durante la terapia de mantenimiento y otros no (ver más adelante). Es imprescindible un control minucioso de los niños durante el mantenimiento con el fin de evaluar la toxicidad farmacológica y el cumplimiento de la quimioterapia oral durante la terapia de mantenimiento. En un protocolo realizado por el COG se indicó que existen diferencias significativas en el cumplimiento de los regímenes de mercaptopurina oral entre varios grupos raciales y socioeconómicos y que el grado de adhesión al tratamiento afecta el riesgo de recaída.
Antes, la práctica generalizada era la administración oral de mercaptopurina en la noche, a partir de la evidencia de estudios antiguos que indicaba que esta práctica mejoraba la SSC. Sin embargo, en un estudio dirigido por el grupo NOPHO, en el que se registró de manera prospectiva la información de ingesta oral, el momento de administración de la mercaptopurina (noche vs. otro momento del día) no tuvo importancia pronóstica. En un estudio del COG, la administración de mercaptopurina a diferentes horas del día en lugar de hacerlo siempre durante la noche se relacionó con tasas más altas de incumplimiento terapéutico. Sin embargo, entre los pacientes cumplidores (es decir, aquellos que tomaron >95 % de las dosis indicadas), no hubo relación entre la hora de la toma de la mercaptopurina y el riesgo de recaída.
Algunos pacientes exhiben toxicidad hematopoyética grave cuando reciben dosis convencionales de mercaptopurina debido a una deficiencia hereditaria (mutación homocigótica) de la tiopurina–S-metiltransferasa, una enzima que inactiva la mercaptopurina. Estos pacientes solo toleran la mercaptopurina en dosis mucho más bajas que las habituales. Los pacientes heterocigóticos para la variante por lo general toleran la mercaptopurina sin efectos tóxicos graves, pero necesitan reducciones de dosis más frecuentes que los pacientes homocigóticos para el alelo normal debido a toxicidad hematopoyética. Los polimorfismos en el gen NUDT15, que se observan más a menudo en pacientes con origen en el este de Asia e hispanos, se han vinculado con una sensibilidad intensa a los efectos mielodepresores de la mercaptopurina.
Evidencia (terapia de mantenimiento):
- En un metanálisis de ensayos aleatorizados se compararon tiopurinas y se estableció lo siguiente.
- La tioguanina no mejoró la SSC general, aunque subgrupos específicos se benefician de su uso.
- El uso continuo de tioguanina en lugar de mercaptopurina durante la fase de mantenimiento se relaciona con un aumento de riesgo de complicaciones hepáticas, como la enfermedad venooclusiva (síndrome de obstrucción sinusoidal) y la hipertensión portal.
- Debido al aumento de la toxicidad de la tioguanina, la mercaptopurina continúa siendo el fármaco estándar de preferencia.
- En el estudio del COG AALL0932 (NCT01190930), pacientes de riesgo estándar del NCI con características de riesgo promedio se asignaron al azar para recibir metotrexato semanal por vía oral durante el mantenimiento en una de las siguientes 2 dosis iniciales: 20 mg/m2 (estándar) o 40 mg/m2 (experimental).[Nivel de evidencia A1]
- No hubo ninguna diferencia significativa en la SSE a 5 años desde el inicio de la terapia de mantenimiento entre los 2 grupos de tratamiento (tasa de SSE a 5 años, 95,1 % en los pacientes que recibieron la dosis estándar vs. 94,2 % en los pacientes que recibieron la dosis experimental; P = 0,92), lo que indica que no hay ningún beneficio con la dosis más alta de metotrexato oral.
- En varios ensayos clínicos del SJCRH y otros grupos, se evaluó un régimen de mantenimiento intensificado con díadas de fármacos en rotación, entre ellos ciclofosfamida y epipodofilotoxinas, así como otros fármacos de mantenimiento más comunes.
- El mantenimiento intensificado con díadas de fármacos en rotación se relacionó con más casos de neutropenia febril y un riesgo más alto de leucemia mielógena aguda secundaria, en especial, cuando se usan epipodofilotoxinas.
A partir de estos hallazgos, el SJCRH modificó los fármacos que se usan en el programa de díadas en rotación durante la fase de mantenimiento. En el ensayo Total XV, los pacientes de riesgo estándar y de riesgo alto recibieron 3 díadas en rotación (mercaptopurina con metotrexato, ciclofosfamida con citarabina y dexametasona con vincristina) durante esta fase de tratamiento. Los pacientes de riesgo bajo recibieron mantenimiento estándar adicional (sin ciclofosfamida ni citarabina).
- En un estudio aleatorizado que se hizo en Argentina, no se demostró ningún beneficio con el abordaje de intensificación, en comparación con un régimen de mantenimiento más estándar, en los pacientes que reciben fases de inducción y consolidación según el tratamiento de base del BFM.
- El mantenimiento intensificado con díadas de fármacos en rotación se relacionó con más casos de neutropenia febril y un riesgo más alto de leucemia mielógena aguda secundaria, en especial, cuando se usan epipodofilotoxinas.
Dosis altas repetidas de vincristina y corticoesteroides
A menudo se añaden dosis altas repetidas de vincristina y corticoesteroides al tratamiento de mantenimiento estándar, aunque el beneficio de estas dosis en el contexto de los regímenes de quimioterapia multifarmacológica contemporáneos genera controversia.
Evidencia (dosis altas repetidas de vincristina y corticoesteroides):
- En un ensayo aleatorizado del CCG que se llevó a cabo en los años ochenta, se demostró mejora del desenlace en los pacientes que recibieron dosis altas repetidas de vincristina y prednisona una vez al mes.
- En un metanálisis, en el que se combinaron los datos de 6 ensayos clínicos de la misma era de tratamiento, se observó que las dosis altas repetidas de vincristina y prednisona ofrecían una ventaja en la SSC. Sin embargo, la SSC general en estos ensayos fue más baja que la observada con el uso de regímenes más contemporáneos.
- En una revisión sistemática de los ensayos clínicos más recientes sobre el efecto de las dosis altas repetidas de vincristina y corticoesteroides, surgió el interrogante de si esas dosis son útiles para el tratamiento actual de la LLA, que incluye una fase temprana más intensiva y la estratificación del riesgo usando la respuesta temprana (ERM) y los factores biológicos.
- En un ensayo aleatorizado multicéntrico de niños con LLA de riesgo intermedio tratados con un régimen del BFM, no se observó ningún beneficio de la adición de 6 dosis altas repetidas de vincristina y dexametasona durante la fase de continuación, aunque las dosis se administraron con menos frecuencia que en otros ensayos que demostraron beneficios.
- En un ensayo multicéntrico pequeño con pacientes de riesgo estándar, se demostró una SSC superior en los pacientes que recibían dosis altas repetidas de vincristina y corticoesteroides. En este estudio no se observó ninguna diferencia en el resultado según el tipo de corticoesteroide (prednisona vs. dexametasona).[Nivel de evidencia A1]
- En el estudio del COG AALL0932 (NCT01190930) , pacientes de riesgo estándar se asignaron al azar durante el mantenimiento para recibir dosis altas repetidas de vincristina y dexametasona cada 4 semanas o cada 12 semanas.[Nivel de evidencia A1]
- En los pacientes de riesgo estándar que se asignaron al azar, la tasa de SSE a 5 años desde el comienzo del mantenimiento fue del 94,6 %. No hubo ninguna diferencia significativa entre los grupos que recibieron el tratamiento cada 4 semanas y cada 12 semanas.
- El Chinese Children’s Cancer Group realizó un ensayo de ausencia de inferioridad para determinar si las dosis altas repetidas de vincristina y dexametasona se podían suprimir durante el segundo año de terapia de mantenimiento. Un año después del inicio de la terapia, 5054 pacientes con LLA sin la fusión BCR::ABL1 (LLA-B y LLA-T, edades 0–18 años) se asignaron al azar para recibir durante el segundo año de quimioterapia de mantenimiento dosis altas repetidas de vincristina y dexametasona cada 8 semanas (7 en total) o a no recibir estas dosis altas repetidas. La ausencia de inferioridad se definió mediante el cálculo del límite superior de confianza unilateral del 95 % de la diferencia en la probabilidad de ERM entre los grupos para garantizar que se descartara una disminución de la SSC del 5 % o más.
- En los pacientes de riesgo bajo (LLA-B de riesgo estándar del NCI con hiperdiploidía alta o ETV6::RUNX1 y ERM baja al final de la inducción), la diferencia en la SSC entre los grupos cumplió con la definición del protocolo de ausencia de inferioridad, lo que indica que al suprimir las dosis altas repetidas de vincristina y dexametasona durante el segundo año de mantenimiento no se produjo una reducción de la SSC superior al 5 %.
- En los pacientes de riesgo intermedio y riesgo alto, la diferencia de SSC a 5 años entre los grupos no cumplió con la definición del protocolo de ausencia de inferioridad (el límite superior de confianza del 95 % para la diferencia fue de 0,055, lo que supera el margen de ausencia de inferioridad de 0,05); por lo tanto, no fue posible concluir que las dosis altas repetidas de vincristina y dexametasona se pudiesen suprimir en estos pacientes sin que se produjera una disminución de la SSC superior al 5 %.
- En una revisión sistemática y un metanálisis se evaluó el efecto de reducir los pulsos de vincristina y esteroides sobre la SSC, la SG y la toxicidad en pacientes con LLA-B. Se examinaron 25 publicaciones que incluyeron más de 12 000 pacientes.
- En este estudio se demostró que los beneficios de estos pulsos observados en ensayos del pasado no se observaban en los ensayos contemporáneos.
- Sin embargo, hubo mayor riesgo de toxicidad no hepática de grado 3+ en el grupo de frecuencia de pulso alta.
- Los autores determinaron que es probable que la reducción o eliminación de pulsos no afecte la supervivencia ni el riesgo de recaída, pero se relaciona con una menor toxicidad.
En varios estudios se ha abordado la pregunta sobre qué tipo de corticoesteroide (dexametasona o prednisona) se debe usar en los regímenes que incluyen dosis altas repetidas de vincristina y corticoesteroides. En estos estudios se indica que la dexametasona se relaciona con una SSC superior, pero también con una mayor frecuencia de complicaciones por uso de corticoesteroides, como toxicidad ósea e infecciones, en especial, en niños de más edad y adolescentes. Al compararse con la prednisona, la dexametasona también se relacionó con una frecuencia más alta de problemas de comportamiento. En un ensayo aleatorizado con 50 pacientes de 3 a 16 años de edad que recibieron quimioterapia de mantenimiento, la administración simultánea de hidrocortisona (dosis fisiológicas) durante las dosis altas repetidas de dexametasona redujo la frecuencia de dificultades del comportamiento, inestabilidad emocional y alteraciones del sueño.
Evidencia (dexametasona vs. prednisona):
- En un estudio del CCG se comparó la dexametasona con la prednisona durante las fases de inducción y mantenimiento en niños de 1 a menos de 10 años de edad con LLA de riesgo más bajo.
- Los pacientes que se asignaron al azar a recibir dexametasona presentaron un número significativamente más bajo de recaídas en el SNC y una tasa de SSC significativamente superior.
- En un ensayo del Medical Research Council (MRC) y el United Kingdom Acute Lymphoblastic Leukaemia (UKALL) se comparó la dexametasona con la prednisolona durante las fases de inducción y mantenimiento en pacientes de riesgo estándar y de riesgo alto.
- La SSC y la incidencia de las recaídas en el SNC y fuera del SNC mejoraron con el uso de la dexametasona.
- La dexametasona se relacionó con un aumento del riesgo de toxicidad por corticoesteroides, como problemas de comportamiento, miopatía y osteopenia.
- En un ensayo del DFCI ALL Consortium, los pacientes se asignaron al azar a recibir dexametasona o prednisona durante todas las fases del tratamiento de posinducción.
- La dexametasona se relacionó con una SSC superior, pero también con una frecuencia más alta de infecciones (en especial, bacteriemia) y, en pacientes de 10 años o más, con una mayor incidencia de osteonecrosis y fracturas.
El beneficio de la dexametasona en los niños de 10 a 18 años exige investigación adicional debido al aumento del riesgo de osteonecrosis por corticoesteroides en este grupo etario.
Duración de la terapia de mantenimiento
Por lo general, la quimioterapia de mantenimiento se prolonga durante 2 a 3 años de RC continua. En algunos estudios, los niños se tratan durante más tiempo que las niñas. En otros ensayos no hay diferencia en la duración del tratamiento según el sexo. No está claro si la prolongación de la terapia de mantenimiento disminuye la recaída en los varones, en especial, en el contexto del tratamiento vigente.[Nivel de evidencia B4] La prolongación de la terapia de mantenimiento por más de 3 años no mejora el desenlace.
Cumplimiento con los medicamentos orales durante la terapia de mantenimiento
El incumplimiento del tratamiento con mercaptopurina durante la terapia de mantenimiento se relaciona con un riesgo de recaída significativo. Se ha desarrollado un modelo de riesgo para predecir qué pacientes presentan un riesgo alto de incumplimiento terapéutico.
Evidencia (cumplimiento terapéutico):
- En el ensayo del COG AALL03N1 (NCT00268528) se estudió el efecto del incumplimiento del tratamiento con mercaptopurina durante la terapia de mantenimiento en 327 niños y adolescentes (169 pacientes hispanos y 158 pacientes blancos no hispanos).
- Se observó un aumento progresivo de la recaída a medida que disminuyó el cumplimiento del tratamiento con mercaptopurina; los CRI oscilaron entre el 4,0 % y el 5,7 % para tasas de cumplimiento que oscilaron entre el 94,9 % y el 90 %, el 89,9 % y el 85 %, y de menos del 85 %. Después del ajuste por otros factores pronósticos (como el grupo de riesgo del NCI y la presencia de anomalías cromosómicas), se observó un aumento progresivo de la recaída de manera inversa a la disminución del cumplimiento del tratamiento con mercaptopurina. No se contaba con los datos de ERM para esta población del estudio, de manera que no se incluyeron en el análisis de factores pronósticos.
- El cumplimiento fue significativamente inferior en los pacientes hispanos, los pacientes mayores de 12 años y los hijos de madres solteras. Sin embargo, entre los pacientes cumplidores, el origen étnico hispano fue un factor de predicción independiente de desenlace adverso.
- En el ensayo AALL03N1 también se incluyeron 71 pacientes estadounidenses de origen asiático y 68 pacientes afroamericanos, además de los pacientes blancos no hispanos e hispanos descritos anteriormente.
- El 20,5 % de los participantes fueron incumplidores cuando se definió el incumplimiento como una tasa de cumplimiento inferior al 90 %.
- Esta tasa de cumplimiento inferior al 90 % se relacionó con aumento del riesgo de recaída (CRI, 3,9).
- Las tasas de cumplimiento fueron significativamente más bajas en pacientes estadounidenses de origen asiático y afroamericanos que en los pacientes blancos no hispanos.
- En una tercer publicación del estudio AALL03N1 se realizaron las siguientes observaciones clave:
- El incumplimiento del tratamiento con mercaptopurina (definido como una tasa de cumplimiento medio <95 %) se tradujo en un aumento de 2,7 veces el riesgo de recaída en comparación con el cumplimiento terapéutico.
- En los pacientes cumplidores, una variabilidad intraindividual alta en las concentraciones de tioguanina (por diferencias en la intensidad de las dosis y las interrupciones del tratamiento) se relacionó con un aumento del riesgo de recaída.
- Los autores de los estudios mencionados antes también encontraron que la autonotificación no fue una medida confiable del cumplimiento terapéutico porque el 84 % de los pacientes, en algún momento, sobrenotificaron el cumplimiento de la toma de mercaptopurina. Los datos indican que se necesita otra medida del cumplimiento además de la autonotificación.
- En otra publicación del estudio AALL03N1, se describieron los hábitos de ingestión de la mercaptopurina, las concentraciones del nucleótido tioguanina (TGN) en los glóbulos rojos, el cumplimiento terapéutico y el riesgo de recaída.
- En los resultados se observó que ciertos hábitos de ingestión (por ejemplo, tomar el medicamento con lácteos o tomarlo a diferentes horas del día) se relacionaban con incumplimiento terapéutico. Sin embargo, después de ajustar por el cumplimiento terapéutico y otros factores pronósticos, los hábitos de ingestión no se relacionaron con el riesgo de recaída.
- En los pacientes cumplidores, no se encontró ninguna relación entre las concentraciones de TGN y los hábitos de ingestión.
- Los autores concluyeron que las restricciones comunes de la ingestión de mercaptopurina no tienen efecto en el desenlace, pero quizás alteren el cumplimiento terapéutico.
Opciones de tratamiento en evaluación clínica
La asignación del tratamiento según el riesgo es una estrategia terapéutica clave que se utiliza en los niños con LLA, y los protocolos están diseñados para poblaciones de pacientes específicas con diferentes grados de riesgo de fracaso del tratamiento. En la sección de este resumen Asignación del tratamiento según el riesgo se describen las características clínicas y de laboratorio que se usan para la estratificación inicial de los niños con LLA en grupos de tratamiento según el riesgo.
La información en inglés sobre los ensayos clínicos patrocinados por el NCI se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presentan ejemplos de ensayos clínicos nacionales o institucionales en curso:
Estudios del COG para la leucemia linfoblástica aguda de células B
Leucemia linfoblástica aguda de riesgo estándar
- COG-AALL1731 (NCT03914625) (A Phase III Trial Investigating Blinatumomab in Combination with Chemotherapy in Patients with Newly Diagnosed Standard Risk or Down Syndrome B-ALL and the Treatment of Patients with Localized B-Lymphoblastic Lymphoma): este protocolo está abierto para pacientes con LLA-B de riesgo estándar del NCI sin síndrome de Down y todos los pacientes con LLA-B y síndrome de Down (<31 años de edad) sin importar el recuento de GB en la presentación inicial. Con el protocolo se evalúa si la adición del anticuerpo biespecífico que se une a las células T, blinatumomab, mejora el desenlace y si la reducción de la duración del tratamiento en los varones (de 3 años desde el inicio de la primera fase de mantenimiento provisional a 2 años desde el inicio de esa fase) afectará de manera adversa la SSE.
Todos los pacientes reciben una inducción de 3 fármacos con dexametasona (sin antraciclinas). Después de terminar la inducción, los pacientes se clasifican en 3 grupos según las mediciones de respuesta biológica y respuesta temprana.
- Riesgo estándar favorable: presencia de ETV6::RUNX1 o trisomía doble (cromosomas 4 y 10), ERM en la sangre periférica <1 % en el día 8, y ERM en la médula ósea <0,01 % en el día 29.
- Riesgo estándar promedio: características biológicas favorables con ERM en la sangre periférica >1 % en el día 8 (pero ERM en la médula ósea <0,01 % en el día 29); o presencia de trisomía doble y ERM en la médula ósea ≥0,01 %, pero <0,1 % en el día 29; o características citogenéticas neutras con ERM en la médula ósea <0,01 % en el día 29.
- Riesgo estándar alto: presencia de ETV6::RUNX1 o características citogenéticas neutras y ERM en la médula ósea ≥0,01 % en el día 29; o presencia de trisomía doble y ERM ≥0,1 % en el día 29; o presencia de características citogenéticas neutras y SCN2 en el momento del diagnóstico, sin importar las medidas de respuesta temprana ni la presencia de características citogenéticas desfavorables (iAMP21, reordenamiento de KMT2A, hipodiploidía (<44 cromosomas) o TCF3::HLF (t(17;19)).
Los pacientes de riesgo estándar favorable recibirán el tratamiento estándar.
A todos los pacientes de riesgo estándar se les medirá la ERM en el día 29 de la inducción mediante una prueba de secuenciación de alto rendimiento (HTS) para la ERM. Los pacientes con ERM indetectable en la HTS recibirán el tratamiento estándar, mientras que los pacientes con ERM detectable en la HTS (o si el resultado de la ERM en la HTS es indeterminado o no está disponible) así como aquellos con trisomía doble y ERM en la médula ósea del ≥0,01 % al <0,1 % en el día 29 serán aptos para participar en una aleatorización y recibir el tratamiento estándar, o el tratamiento estándar con 2 ciclos de blinatumomab.
Los pacientes de riesgo estándar alto recibirán el tratamiento de base del BFM intensificado (riesgo alto del NCI). Todos los pacientes con una ERM >1 % al final de la consolidación salen del protocolo de tratamiento. Los pacientes con una ERM <0,1 % al final de la consolidación serán aptos para la aleatorización al tratamiento de base para el grupo de riesgo alto del NCI solo o combinado con 2 ciclos de blinatumomab. Los pacientes con una ERM del ≥0,1 % al <1 % al final de la consolidación se asignarán de manera directa a recibir el tratamiento de base para el grupo de riesgo alto del NCI con 2 ciclos de blinatumomab.
Los pacientes con síndrome de Down y riesgo estándar del NCI que cumplen con la definición de riesgo estándar promedio recibirán el mismo tratamiento que los pacientes sin síndrome de Down del grupo de riesgo estándar promedio, según se describió antes. El resto de los pacientes con síndrome de Down, incluso los de riesgo alto del NCI, características biológicas desfavorables y ERM alta el día 29 se clasificarán en el grupo de síndrome de Down de riesgo alto y se asignarán al azar a recibir 2 ciclos de blinatumomab, además del régimen de quimioterapia reducido en el que se omiten los elementos intensivos del tratamiento de base del BFM intensificado. Los elementos que se omiten son las antraciclinas durante la inducción y la quimioterapia a base de ciclofosfamida y citarabina durante la segunda parte de la intensificación diferida.
El tratamiento durará lo mismo para todos los pacientes (2 años desde el comienzo de la primera fase de mantenimiento provisional) sin importar el grupo de riesgo. Esto representa una reducción de 1 año en la duración del tratamiento para los varones, en comparación con el tratamiento estándar del COG.
Leucemia linfoblástica aguda de riesgo alto y riesgo muy alto
- COG-AALL1732 (NCT03959085) (A Phase III Randomized Trial of Inotuzumab Ozogamicin for Newly Diagnosed High-Risk B-ALL; Risk-Adapted Postinduction Therapy for High-Risk B-ALL, Mixed Phenotype Acute Leukemia [MPAL], and Disseminated B-Lymphoblastic Lymphoma): este protocolo está abierto para pacientes con LLA de riesgo alto del NCI sin síndrome de Down, cualquier paciente con LAFM y pacientes con linfoma linfoblástico de células B diseminado. Los pacientes con LLA-B de riesgo estándar del NCI que recibieron tratamiento previo con corticoesteroides, exhiben un estado SNC3 o enfermedad testicular en el momento del diagnóstico, también reúnen las condiciones para participar en este estudio.
En los pacientes con LLA-B del protocolo se evalúa si la adición de 2 bloques de inotuzumab ozogamicina a un tratamiento de base del BFM modificado mejorará la SSE, además se prueba si la reducción de la duración del tratamiento en los varones (de 3 años desde el inicio de la primera fase de mantenimiento provisional a 2 años desde el inicio de esta fase) afectará de manera adversa la SSE. Asimismo, en el estudio se procura determinar la SSC de los pacientes con LAFM y linfoma linfoblástico de células B diseminado que reciben tratamiento con un régimen quimioterapéutico para la LLA de riesgo estándar alto.
Todos los pacientes reciben una inducción de 4 fármacos (que incluye prednisona y daunorrubicina). Al finalizar la inducción, el tratamiento posterior depende de la edad, las características biológicas y la respuesta al tratamiento.
- Riesgo alto favorable: los pacientes menores de 10 años con fusiones ETV6::RUNX1 o hiperdiploidía alta con trisomías de los cromosomas 4 y 10 y que alcanzan una ERM <0,01 % al final de la inducción recibirán un régimen del BFM modificado con una fase de mantenimiento provisional (dosis altas de metotrexato), y no se someten a aleatorización.
- Otros pacientes con LLA-B de riesgo alto que no satisfacen los criterios de riesgo alto favorable, pero que alcanzan una ERM <0,01 % (grupo de riesgo alto del NCI) o <1 % (grupo de riesgo estándar del NCI) al final de la consolidación, serán aptos para la aleatorización al tratamiento del BFM modificado con 2 bloques de inotuzumab o sin ellos. Los pacientes que obtienen un resultado negativo para CD22 en el momento del diagnóstico (o que tienen estado desconocido de CD22) no reúnen las condiciones para la aleatorización y se retiran del protocolo de tratamiento.
- Los pacientes con LAFM y linfoma linfoblástico de células B diseminado recibirán un tratamiento de base del BFM modificado con 2 fases de mantenimiento provisional, pero no reúnen las condiciones para la aleatorización.
El tratamiento durará lo mismo para todos los pacientes (2 años desde el comienzo de la primera fase de mantenimiento provisional). Esto representa una reducción de 1 año en la duración del tratamiento para los varones, en comparación con el tratamiento estándar. Los pacientes con LLA-B de riesgo alto del NCI con ERM al final de la consolidación ≥0,01 % se retiran del protocolo de tratamiento y reúnen las condiciones para inscribirse en el ensayo de COG-AALL1721 (mencionado antes). Los pacientes de riesgo estándar del NCI con ERM al final de la consolidación ≥1 % se retiran del protocolo de tratamiento y no reúnen las condiciones para inscribirse en el ensayo de COG-AALL1721.
- COG-AALL1721 (NCT03876769) (Study of Efficacy and Safety of Tisagenlecleucel in High-Risk B-ALL End-of-Consolidation MRD-Positive Patients): este protocolo está abierto a pacientes con LLA-B de riesgo alto del NCI de 1 a 25 años de edad, RC en el estudio morfológico al final de la inducción y ERM al final de la consolidación ≥0,01 %. El objetivo principal del ensayo es evaluar la eficacia del tisagenlecleucel (células T con receptor de antígeno quimérico [CAR] dirigido a CD19) como tratamiento definitivo en esta población de pacientes, en particular con el fin de determinar si la tasa de SSE a 5 años del tisagenlecleucel supera el 55 %.
Los pacientes inscritos en este ensayo se someterán a leucocitaféresis para obtener células T autógenas que se enviarán para la producción del tisagenlecleucel. Mientras se espera que termine la producción, los pacientes iniciarán la primera fase de mantenimiento provisional (dosis altas de metotrexato); es posible que esta fase se interrumpa tan pronto como se disponga del producto. Una vez disponible el tisagenlecleucel, los pacientes recibirán quimioterapia linfocitorreductora e infusión de tisagenlecleucel. Después del tisagenlecleucel no se administrarán otros tratamientos para la leucemia. En el día 29 después de la administración del tisagenlecleucel se empezarán a obtener muestras de médula a intervalos regulares con el fin de evaluar el estado de la enfermedad; además se examinarán muestras de sangre periférica para detectar indicios de aplasia de células B.
Para inscribirse en este ensayo, los pacientes deben contar con un resultado positivo para CD19 en el momento del diagnóstico. Se excluyen los pacientes con médula de tipo M3 al final de la inducción, médula de tipo M2 o M3 al final de la consolidación, hipodiploidía (<44 cromosomas), LLA BCR::ABL1, o que hayan recibido antes tratamiento con un inhibidor de tirosina–cinasas.
- AALL1631 (NCT03007147) (Imatinib Mesylate and Combination Chemotherapy in Treating Patients With Newly Diagnosed BCR::ABL1 [Ph+] ALL): AALL1631 es un protocolo colaborativo internacional dirigido por el COG y el grupo europeo EsPhALL. Los pacientes con LLA similar a BCR::ABL1 y fusiones de clase ABL (definidas como aquellas que afectan ABL1, ABL2, CSF1R, PDGFRB, o PDGFRA) son aptos para la inscripción en el estudio. Estos pacientes se incorporaron al ensayo después de completar el primer mes de tratamiento (inducción IA) y reciben quimioterapia combinada con imatinib (se permite el pretratamiento con imatinib durante la inducción IA). Después de la fase de inducción IB (semanas 10–12), se evalúa la ERM mediante PCR para la inmunoglobulina H y el receptor de células T (IgH-TCR), de manera que los pacientes se clasifican como de riesgo estándar (ERM <0,05 %) o riesgo alto (ERM >0,05 %). Los pacientes de riesgo estándar se asignan al azar a recibir uno de los siguientes regímenes de quimioterapia citotóxica de base:
- El tratamiento de base de EsPhALL utilizado en los protocolos anteriores de EsPhALL y COG AALL1122; o
- Un régimen menos intensivo similar a los que se suelen administrar a pacientes con LLA-B de riesgo alto que no presentan BCR::ABL1 en los ensayos del COG.
Los pacientes de riesgo estándar de ambos grupos continuarán recibiendo imatinib hasta que completen toda la quimioterapia prevista (2 años de tratamiento). El objetivo de la aleatorización al grupo de riesgo estándar es determinar si la quimioterapia de base menos intensiva se relaciona con una SSE similar y tasas más bajas de morbilidad y mortalidad relacionada con el tratamiento en comparación con la terapia estándar (quimioterapia de base de EsPhALL).
Los pacientes de riesgo alto se someterán a un TCMH después de completar 3 bloques de consolidación de quimioterapia. El tratamiento con imatinib se reiniciará después del TCMH y se administrará desde el día 56 hasta el día 365. El objetivo es evaluar la viabilidad de la administración de imatinib posterior al TCMH y describir los desenlaces de estos pacientes.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
References
- Möricke A, Zimmermann M, Reiter A, et al.: Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 24 (2): 265-84, 2010.
- Pui CH, Pei D, Sandlund JT, et al.: Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 24 (2): 371-82, 2010.
- Silverman LB, Stevenson KE, O'Brien JE, et al.: Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia 24 (2): 320-34, 2010.
- Oudot C, Auclerc MF, Levy V, et al.: Prognostic factors for leukemic induction failure in children with acute lymphoblastic leukemia and outcome after salvage therapy: the FRALLE 93 study. J Clin Oncol 26 (9): 1496-503, 2008.
- Salzer WL, Devidas M, Carroll WL, et al.: Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984-2001: a report from the children's oncology group. Leukemia 24 (2): 355-70, 2010.
- Bostrom BC, Sensel MR, Sather HN, et al.: Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: a report from the Children's Cancer Group. Blood 101 (10): 3809-17, 2003.
- Mitchell CD, Richards SM, Kinsey SE, et al.: Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomized trial. Br J Haematol 129 (6): 734-45, 2005.
- Larsen EC, Devidas M, Chen S, et al.: Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults With High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group Study AALL0232. J Clin Oncol 34 (20): 2380-8, 2016.
- Möricke A, Zimmermann M, Valsecchi MG, et al.: Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood 127 (17): 2101-12, 2016.
- McNeer JL, Nachman JB: The optimal use of steroids in paediatric acute lymphoblastic leukaemia: no easy answers. Br J Haematol 149 (5): 638-52, 2010.
- Silverman LB, Supko JG, Stevenson KE, et al.: Intravenous PEG-asparaginase during remission induction in children and adolescents with newly diagnosed acute lymphoblastic leukemia. Blood 115 (7): 1351-3, 2010.
- Rizzari C, Citterio M, Zucchetti M, et al.: A pharmacological study on pegylated asparaginase used in front-line treatment of children with acute lymphoblastic leukemia. Haematologica 91 (1): 24-31, 2006.
- Place AE, Stevenson KE, Vrooman LM, et al.: Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): a randomised, open-label phase 3 trial. Lancet Oncol 16 (16): 1677-90, 2015.
- Asselin BL, Whitin JC, Coppola DJ, et al.: Comparative pharmacokinetic studies of three asparaginase preparations. J Clin Oncol 11 (9): 1780-6, 1993.
- Avramis VI, Sencer S, Periclou AP, et al.: A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: a Children's Cancer Group study. Blood 99 (6): 1986-94, 2002.
- Tram Henriksen L, Gottschalk Højfeldt S, Schmiegelow K, et al.: Prolonged first-line PEG-asparaginase treatment in pediatric acute lymphoblastic leukemia in the NOPHO ALL2008 protocol-Pharmacokinetics and antibody formation. Pediatr Blood Cancer 64 (12): , 2017.
- Schore RJ, Devidas M, Bleyer A, et al.: Plasma asparaginase activity and asparagine depletion in acute lymphoblastic leukemia patients treated with pegaspargase on Children's Oncology Group AALL07P4. Leuk Lymphoma 60 (7): 1740-1748, 2019.
- Jeha S, Pei D, Choi J, et al.: Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J Clin Oncol 37 (35): 3377-3391, 2019.
- Kloos RQH, Pieters R, Jumelet FMV, et al.: Individualized Asparaginase Dosing in Childhood Acute Lymphoblastic Leukemia. J Clin Oncol 38 (7): 715-724, 2020.
- Gupta S, Wang C, Raetz EA, et al.: Impact of Asparaginase Discontinuation on Outcome in Childhood Acute Lymphoblastic Leukemia: A Report From the Children's Oncology Group. J Clin Oncol 38 (17): 1897-1905, 2020.
- van der Sluis IM, Vrooman LM, Pieters R, et al.: Consensus expert recommendations for identification and management of asparaginase hypersensitivity and silent inactivation. Haematologica 101 (3): 279-85, 2016.
- Bleyer A, Asselin BL, Koontz SE, et al.: Clinical application of asparaginase activity levels following treatment with pegaspargase. Pediatr Blood Cancer 62 (6): 1102-5, 2015.
- Tong WH, Pieters R, Kaspers GJ, et al.: A prospective study on drug monitoring of PEGasparaginase and Erwinia asparaginase and asparaginase antibodies in pediatric acute lymphoblastic leukemia. Blood 123 (13): 2026-33, 2014.
- Vrooman LM, Stevenson KE, Supko JG, et al.: Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol 31 (9): 1202-10, 2013.
- Gottschalk Højfeldt S, Grell K, Abrahamsson J, et al.: Relapse risk following truncation of pegylated asparaginase in childhood acute lymphoblastic leukemia. Blood 137 (17): 2373-2382, 2021.
- van der Sluis IM, Brigitha LJ, Fiocco M, et al.: Continuous PEGasparaginase Dosing Reduces Hypersensitivity Reactions in Pediatric ALL: A Dutch Childhood Oncology Group ALL11 Randomized Trial. J Clin Oncol 42 (14): 1676-1686, 2024.
- Li RJ, Jin R, Liu C, et al.: FDA Approval Summary: Calaspargase Pegol-mknl For Treatment of Acute Lymphoblastic Leukemia in Children and Young Adults. Clin Cancer Res 26 (2): 328-331, 2020.
- Angiolillo AL, Schore RJ, Devidas M, et al.: Pharmacokinetic and pharmacodynamic properties of calaspargase pegol Escherichia coli L-asparaginase in the treatment of patients with acute lymphoblastic leukemia: results from Children's Oncology Group Study AALL07P4. J Clin Oncol 32 (34): 3874-82, 2014.
- Vrooman LM, Blonquist TM, Stevenson KE, et al.: Efficacy and Toxicity of Pegaspargase and Calaspargase Pegol in Childhood Acute Lymphoblastic Leukemia: Results of DFCI 11-001. J Clin Oncol 39 (31): 3496-3505, 2021.
- Salzer WL, Asselin B, Supko JG, et al.: Erwinia asparaginase achieves therapeutic activity after pegaspargase allergy: a report from the Children's Oncology Group. Blood 122 (4): 507-14, 2013.
- Vrooman LM, Kirov II, Dreyer ZE, et al.: Activity and Toxicity of Intravenous Erwinia Asparaginase Following Allergy to E. coli-Derived Asparaginase in Children and Adolescents With Acute Lymphoblastic Leukemia. Pediatr Blood Cancer 63 (2): 228-33, 2016.
- Maese L, Loh ML, Choi MR, et al.: Recombinant Erwinia asparaginase (JZP458) in acute lymphoblastic leukemia: results from the phase 2/3 AALL1931 study. Blood 141 (7): 704-712, 2023.
- Escherich G, Zimmermann M, Janka-Schaub G, et al.: Doxorubicin or daunorubicin given upfront in a therapeutic window are equally effective in children with newly diagnosed acute lymphoblastic leukemia. A randomized comparison in trial CoALL 07-03. Pediatr Blood Cancer 60 (2): 254-7, 2013.
- Pui CH, Sandlund JT, Pei D, et al.: Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children's Research Hospital. Blood 104 (9): 2690-6, 2004.
- Schrappe M, Reiter A, Ludwig WD, et al.: Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 95 (11): 3310-22, 2000.
- Moghrabi A, Levy DE, Asselin B, et al.: Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood 109 (3): 896-904, 2007.
- Prucker C, Attarbaschi A, Peters C, et al.: Induction death and treatment-related mortality in first remission of children with acute lymphoblastic leukemia: a population-based analysis of the Austrian Berlin-Frankfurt-Münster study group. Leukemia 23 (7): 1264-9, 2009.
- Buchmann S, Schrappe M, Baruchel A, et al.: Remission, treatment failure, and relapse in pediatric ALL: an international consensus of the Ponte-di-Legno Consortium. Blood 139 (12): 1785-1793, 2022.
- Balduzzi A, Valsecchi MG, Uderzo C, et al.: Chemotherapy versus allogeneic transplantation for very-high-risk childhood acute lymphoblastic leukaemia in first complete remission: comparison by genetic randomisation in an international prospective study. Lancet 366 (9486): 635-42, 2005 Aug 20-26.
- Silverman LB, Gelber RD, Young ML, et al.: Induction failure in acute lymphoblastic leukemia of childhood. Cancer 85 (6): 1395-404, 1999.
- Schrappe M, Hunger SP, Pui CH, et al.: Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med 366 (15): 1371-81, 2012.
- Raetz EA, Rebora P, Conter V, et al.: Outcome for Children and Young Adults With T-Cell ALL and Induction Failure in Contemporary Trials. J Clin Oncol 41 (32): 5025-5034, 2023.
- Dunsmore KP, Winter SS, Devidas M, et al.: Children's Oncology Group AALL0434: A Phase III Randomized Clinical Trial Testing Nelarabine in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia. J Clin Oncol 38 (28): 3282-3293, 2020.
- Gaynon PS, Desai AA, Bostrom BC, et al.: Early response to therapy and outcome in childhood acute lymphoblastic leukemia: a review. Cancer 80 (9): 1717-26, 1997.
- Borowitz MJ, Devidas M, Hunger SP, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood 111 (12): 5477-85, 2008.
- Borowitz MJ, Wood BL, Devidas M, et al.: Prognostic significance of minimal residual disease in high risk B-ALL: a report from Children's Oncology Group study AALL0232. Blood 126 (8): 964-71, 2015.
- van Dongen JJ, Seriu T, Panzer-Grümayer ER, et al.: Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 352 (9142): 1731-8, 1998.
- Zhou J, Goldwasser MA, Li A, et al.: Quantitative analysis of minimal residual disease predicts relapse in children with B-lineage acute lymphoblastic leukemia in DFCI ALL Consortium Protocol 95-01. Blood 110 (5): 1607-11, 2007.
- Conter V, Bartram CR, Valsecchi MG, et al.: Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 115 (16): 3206-14, 2010.
- Wood B, Wu D, Crossley B, et al.: Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood 131 (12): 1350-1359, 2018.
- Vora A, Goulden N, Mitchell C, et al.: Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): a randomised controlled trial. Lancet Oncol 15 (8): 809-18, 2014.
- Coustan-Smith E, Sancho J, Behm FG, et al.: Prognostic importance of measuring early clearance of leukemic cells by flow cytometry in childhood acute lymphoblastic leukemia. Blood 100 (1): 52-8, 2002.
- Basso G, Veltroni M, Valsecchi MG, et al.: Risk of relapse of childhood acute lymphoblastic leukemia is predicted by flow cytometric measurement of residual disease on day 15 bone marrow. J Clin Oncol 27 (31): 5168-74, 2009.
- Schrappe M, Valsecchi MG, Bartram CR, et al.: Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood 118 (8): 2077-84, 2011.
- Karsa M, Dalla Pozza L, Venn NC, et al.: Improving the identification of high risk precursor B acute lymphoblastic leukemia patients with earlier quantification of minimal residual disease. PLoS One 8 (10): e76455, 2013.
- Nachman JB, Sather HN, Sensel MG, et al.: Augmented post-induction therapy for children with high-risk acute lymphoblastic leukemia and a slow response to initial therapy. N Engl J Med 338 (23): 1663-71, 1998.
- Seibel NL, Steinherz PG, Sather HN, et al.: Early postinduction intensification therapy improves survival for children and adolescents with high-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 111 (5): 2548-55, 2008.
- Albertsen BK, Grell K, Abrahamsson J, et al.: Intermittent Versus Continuous PEG-Asparaginase to Reduce Asparaginase-Associated Toxicities: A NOPHO ALL2008 Randomized Study. J Clin Oncol 37 (19): 1638-1646, 2019.
- Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.
- Veerman AJ, Hählen K, Kamps WA, et al.: High cure rate with a moderately intensive treatment regimen in non-high-risk childhood acute lymphoblastic leukemia. Results of protocol ALL VI from the Dutch Childhood Leukemia Study Group. J Clin Oncol 14 (3): 911-8, 1996.
- Chauvenet AR, Martin PL, Devidas M, et al.: Antimetabolite therapy for lesser-risk B-lineage acute lymphoblastic leukemia of childhood: a report from Children's Oncology Group Study P9201. Blood 110 (4): 1105-11, 2007.
- Gustafsson G, Kreuger A, Clausen N, et al.: Intensified treatment of acute childhood lymphoblastic leukaemia has improved prognosis, especially in non-high-risk patients: the Nordic experience of 2648 patients diagnosed between 1981 and 1996. Nordic Society of Paediatric Haematology and Oncology (NOPHO) Acta Paediatr 87 (11): 1151-61, 1998.
- Mattano LA, Devidas M, Maloney KW, et al.: Favorable Trisomies and ETV6-RUNX1 Predict Cure in Low-Risk B-Cell Acute Lymphoblastic Leukemia: Results From Children's Oncology Group Trial AALL0331. J Clin Oncol 39 (14): 1540-1552, 2021.
- Mahoney DH, Shuster JJ, Nitschke R, et al.: Intensification with intermediate-dose intravenous methotrexate is effective therapy for children with lower-risk B-precursor acute lymphoblastic leukemia: A Pediatric Oncology Group study. J Clin Oncol 18 (6): 1285-94, 2000.
- Veerman AJ, Kamps WA, van den Berg H, et al.: Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004). Lancet Oncol 10 (10): 957-66, 2009.
- Schore RJ, Angiolillo AJ, Kairalla JA, et al.: Outcomes with reduced intensity therapy in a low-risk subset of children with National Cancer Institute (NCI) standard-risk (SR) B-lymphoblastic leukemia (B-ALL): A report from Children’s Oncology Group (COG) AALL0932. [Abstract] J Clin Oncol 38 (15 suppl): A-10509, 2020. Also available online. Last accessed June 13, 2022.
- Silverman LB, Gelber RD, Dalton VK, et al.: Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood 97 (5): 1211-8, 2001.
- Pession A, Valsecchi MG, Masera G, et al.: Long-term results of a randomized trial on extended use of high dose L-asparaginase for standard risk childhood acute lymphoblastic leukemia. J Clin Oncol 23 (28): 7161-7, 2005.
- Coustan-Smith E, Sancho J, Hancock ML, et al.: Use of peripheral blood instead of bone marrow to monitor residual disease in children with acute lymphoblastic leukemia. Blood 100 (7): 2399-402, 2002.
- Stow P, Key L, Chen X, et al.: Clinical significance of low levels of minimal residual disease at the end of remission induction therapy in childhood acute lymphoblastic leukemia. Blood 115 (23): 4657-63, 2010.
- Gaynon PS, Angiolillo AL, Carroll WL, et al.: Long-term results of the children's cancer group studies for childhood acute lymphoblastic leukemia 1983-2002: a Children's Oncology Group Report. Leukemia 24 (2): 285-97, 2010.
- Riehm H, Gadner H, Henze G, et al.: Results and significance of six randomized trials in four consecutive ALL-BFM studies. Hamatol Bluttransfus 33: 439-50, 1990.
- Hutchinson RJ, Gaynon PS, Sather H, et al.: Intensification of therapy for children with lower-risk acute lymphoblastic leukemia: long-term follow-up of patients treated on Children's Cancer Group Trial 1881. J Clin Oncol 21 (9): 1790-7, 2003.
- Möricke A, Reiter A, Zimmermann M, et al.: Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 111 (9): 4477-89, 2008.
- Matloub Y, Bostrom BC, Hunger SP, et al.: Escalating intravenous methotrexate improves event-free survival in children with standard-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 118 (2): 243-51, 2011.
- Matloub Y, Stork L, Asselin B, et al.: Outcome of Children with Standard-Risk T-Lineage Acute Lymphoblastic Leukemia--Comparison among Different Treatment Strategies. Pediatr Blood Cancer 63 (2): 255-61, 2016.
- Maloney KW, Devidas M, Wang C, et al.: Outcome in Children With Standard-Risk B-Cell Acute Lymphoblastic Leukemia: Results of Children's Oncology Group Trial AALL0331. J Clin Oncol 38 (6): 602-612, 2020.
- Vora A, Goulden N, Wade R, et al.: Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol 14 (3): 199-209, 2013.
- Schrappe M, Bleckmann K, Zimmermann M, et al.: Reduced-Intensity Delayed Intensification in Standard-Risk Pediatric Acute Lymphoblastic Leukemia Defined by Undetectable Minimal Residual Disease: Results of an International Randomized Trial (AIEOP-BFM ALL 2000). J Clin Oncol 36 (3): 244-253, 2018.
- Ariffin H, Chiew EKH, Oh BLZ, et al.: Anthracycline-Free Protocol for Favorable-Risk Childhood ALL: A Noninferiority Comparison Between Malaysia-Singapore ALL 2003 and ALL 2010 Studies. J Clin Oncol 41 (20): 3642-3651, 2023.
- Pui CH, Mahmoud HH, Rivera GK, et al.: Early intensification of intrathecal chemotherapy virtually eliminates central nervous system relapse in children with acute lymphoblastic leukemia. Blood 92 (2): 411-5, 1998.
- Mattano LA, Sather HN, Trigg ME, et al.: Osteonecrosis as a complication of treating acute lymphoblastic leukemia in children: a report from the Children's Cancer Group. J Clin Oncol 18 (18): 3262-72, 2000.
- Aricò M, Valsecchi MG, Conter V, et al.: Improved outcome in high-risk childhood acute lymphoblastic leukemia defined by prednisone-poor response treated with double Berlin-Frankfurt-Muenster protocol II. Blood 100 (2): 420-6, 2002.
- Steinherz PG, Seibel NL, Sather H, et al.: Treatment of higher risk acute lymphoblastic leukemia in young people (CCG-1961), long-term follow-up: a report from the Children's Oncology Group. Leukemia 33 (9): 2144-2154, 2019.
- Mattano LA, Devidas M, Nachman JB, et al.: Effect of alternate-week versus continuous dexamethasone scheduling on the risk of osteonecrosis in paediatric patients with acute lymphoblastic leukaemia: results from the CCG-1961 randomised cohort trial. Lancet Oncol 13 (9): 906-15, 2012.
- Moorman AV, Antony G, Wade R, et al.: Time to Cure for Childhood and Young Adult Acute Lymphoblastic Leukemia Is Independent of Early Risk Factors: Long-Term Follow-Up of the UKALL2003 Trial. J Clin Oncol 40 (36): 4228-4239, 2022.
- Lipshultz SE, Scully RE, Lipsitz SR, et al.: Assessment of dexrazoxane as a cardioprotectant in doxorubicin-treated children with high-risk acute lymphoblastic leukaemia: long-term follow-up of a prospective, randomised, multicentre trial. Lancet Oncol 11 (10): 950-61, 2010.
- Barry EV, Vrooman LM, Dahlberg SE, et al.: Absence of secondary malignant neoplasms in children with high-risk acute lymphoblastic leukemia treated with dexrazoxane. J Clin Oncol 26 (7): 1106-11, 2008.
- Asselin BL, Devidas M, Chen L, et al.: Cardioprotection and Safety of Dexrazoxane in Patients Treated for Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma: A Report of the Children's Oncology Group Randomized Trial Pediatric Oncology Group 9404. J Clin Oncol 34 (8): 854-62, 2016.
- Schultz KR, Pullen DJ, Sather HN, et al.: Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children's Cancer Group (CCG). Blood 109 (3): 926-35, 2007.
- van Binsbergen AL, de Haas V, van der Velden VHJ, et al.: Efficacy and toxicity of high-risk therapy of the Dutch Childhood Oncology Group in childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 69 (2): e29387, 2022.
- Schrauder A, Reiter A, Gadner H, et al.: Superiority of allogeneic hematopoietic stem-cell transplantation compared with chemotherapy alone in high-risk childhood T-cell acute lymphoblastic leukemia: results from ALL-BFM 90 and 95. J Clin Oncol 24 (36): 5742-9, 2006.
- Ribera JM, Ortega JJ, Oriol A, et al.: Comparison of intensive chemotherapy, allogeneic, or autologous stem-cell transplantation as postremission treatment for children with very high risk acute lymphoblastic leukemia: PETHEMA ALL-93 Trial. J Clin Oncol 25 (1): 16-24, 2007.
- Pieters R, de Groot-Kruseman H, Van der Velden V, et al.: Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol 34 (22): 2591-601, 2016.
- Conter V, Valsecchi MG, Parasole R, et al.: Childhood high-risk acute lymphoblastic leukemia in first remission: results after chemotherapy or transplant from the AIEOP ALL 2000 study. Blood 123 (10): 1470-8, 2014.
- Ifversen M, Turkiewicz D, Marquart HV, et al.: Low burden of minimal residual disease prior to transplantation in children with very high risk acute lymphoblastic leukaemia: The NOPHO ALL2008 experience. Br J Haematol 184 (6): 982-993, 2019.
- Pui CH, Rebora P, Schrappe M, et al.: Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Retrospective Multinational Study. J Clin Oncol 37 (10): 770-779, 2019.
- McNeer JL, Devidas M, Dai Y, et al.: Hematopoietic Stem-Cell Transplantation Does Not Improve the Poor Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group. J Clin Oncol 37 (10): 780-789, 2019.
- Bhatia S, Landier W, Shangguan M, et al.: Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the children's oncology group. J Clin Oncol 30 (17): 2094-101, 2012.
- Bhatia S, Landier W, Hageman L, et al.: 6MP adherence in a multiracial cohort of children with acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 124 (15): 2345-53, 2014.
- Schmiegelow K, Glomstein A, Kristinsson J, et al.: Impact of morning versus evening schedule for oral methotrexate and 6-mercaptopurine on relapse risk for children with acute lymphoblastic leukemia. Nordic Society for Pediatric Hematology and Oncology (NOPHO). J Pediatr Hematol Oncol 19 (2): 102-9, 1997 Mar-Apr.
- Clemmensen KK, Christensen RH, Shabaneh DN, et al.: The circadian schedule for childhood acute lymphoblastic leukemia maintenance therapy does not influence event-free survival in the NOPHO ALL92 protocol. Pediatr Blood Cancer 61 (4): 653-8, 2014.
- Landier W, Hageman L, Chen Y, et al.: Mercaptopurine Ingestion Habits, Red Cell Thioguanine Nucleotide Levels, and Relapse Risk in Children With Acute Lymphoblastic Leukemia: A Report From the Children's Oncology Group Study AALL03N1. J Clin Oncol 35 (15): 1730-1736, 2017.
- Relling MV, Hancock ML, Rivera GK, et al.: Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 91 (23): 2001-8, 1999.
- Andersen JB, Szumlanski C, Weinshilboum RM, et al.: Pharmacokinetics, dose adjustments, and 6-mercaptopurine/methotrexate drug interactions in two patients with thiopurine methyltransferase deficiency. Acta Paediatr 87 (1): 108-11, 1998.
- Yang JJ, Landier W, Yang W, et al.: Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol 33 (11): 1235-42, 2015.
- Moriyama T, Nishii R, Perez-Andreu V, et al.: NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet 48 (4): 367-73, 2016.
- Zhou H, Li L, Yang P, et al.: Optimal predictor for 6-mercaptopurine intolerance in Chinese children with acute lymphoblastic leukemia: NUDT15, TPMT, or ITPA genetic variants? BMC Cancer 18 (1): 516, 2018.
- Escherich G, Richards S, Stork LC, et al.: Meta-analysis of randomised trials comparing thiopurines in childhood acute lymphoblastic leukaemia. Leukemia 25 (6): 953-9, 2011.
- Broxson EH, Dole M, Wong R, et al.: Portal hypertension develops in a subset of children with standard risk acute lymphoblastic leukemia treated with oral 6-thioguanine during maintenance therapy. Pediatr Blood Cancer 44 (3): 226-31, 2005.
- De Bruyne R, Portmann B, Samyn M, et al.: Chronic liver disease related to 6-thioguanine in children with acute lymphoblastic leukaemia. J Hepatol 44 (2): 407-10, 2006.
- Vora A, Mitchell CD, Lennard L, et al.: Toxicity and efficacy of 6-thioguanine versus 6-mercaptopurine in childhood lymphoblastic leukaemia: a randomised trial. Lancet 368 (9544): 1339-48, 2006.
- Jacobs SS, Stork LC, Bostrom BC, et al.: Substitution of oral and intravenous thioguanine for mercaptopurine in a treatment regimen for children with standard risk acute lymphoblastic leukemia: a collaborative Children's Oncology Group/National Cancer Institute pilot trial (CCG-1942). Pediatr Blood Cancer 49 (3): 250-5, 2007.
- Stork LC, Matloub Y, Broxson E, et al.: Oral 6-mercaptopurine versus oral 6-thioguanine and veno-occlusive disease in children with standard-risk acute lymphoblastic leukemia: report of the Children's Oncology Group CCG-1952 clinical trial. Blood 115 (14): 2740-8, 2010.
- Angiolillo AL, Schore RJ, Kairalla JA, et al.: Excellent Outcomes With Reduced Frequency of Vincristine and Dexamethasone Pulses in Standard-Risk B-Lymphoblastic Leukemia: Results From Children's Oncology Group AALL0932. J Clin Oncol 39 (13): 1437-1447, 2021.
- Felice MS, Rossi JG, Gallego MS, et al.: No advantage of a rotational continuation phase in acute lymphoblastic leukemia in childhood treated with a BFM back-bone therapy. Pediatr Blood Cancer 57 (1): 47-55, 2011.
- Hijiya N, Hudson MM, Lensing S, et al.: Cumulative incidence of secondary neoplasms as a first event after childhood acute lymphoblastic leukemia. JAMA 297 (11): 1207-15, 2007.
- Pui CH, Ribeiro RC, Hancock ML, et al.: Acute myeloid leukemia in children treated with epipodophyllotoxins for acute lymphoblastic leukemia. N Engl J Med 325 (24): 1682-7, 1991.
- Bleyer WA, Sather HN, Nickerson HJ, et al.: Monthly pulses of vincristine and prednisone prevent bone marrow and testicular relapse in low-risk childhood acute lymphoblastic leukemia: a report of the CCG-161 study by the Childrens Cancer Study Group. J Clin Oncol 9 (6): 1012-21, 1991.
- Duration and intensity of maintenance chemotherapy in acute lymphoblastic leukaemia: overview of 42 trials involving 12 000 randomised children. Childhood ALL Collaborative Group. Lancet 347 (9018): 1783-8, 1996.
- Eden TO, Pieters R, Richards S, et al.: Systematic review of the addition of vincristine plus steroid pulses in maintenance treatment for childhood acute lymphoblastic leukaemia - an individual patient data meta-analysis involving 5,659 children. Br J Haematol 149 (5): 722-33, 2010.
- Conter V, Valsecchi MG, Silvestri D, et al.: Pulses of vincristine and dexamethasone in addition to intensive chemotherapy for children with intermediate-risk acute lymphoblastic leukaemia: a multicentre randomised trial. Lancet 369 (9556): 123-31, 2007.
- De Moerloose B, Suciu S, Bertrand Y, et al.: Improved outcome with pulses of vincristine and corticosteroids in continuation therapy of children with average risk acute lymphoblastic leukemia (ALL) and lymphoblastic non-Hodgkin lymphoma (NHL): report of the EORTC randomized phase 3 trial 58951. Blood 116 (1): 36-44, 2010.
- Yang W, Cai J, Shen S, et al.: Pulse therapy with vincristine and dexamethasone for childhood acute lymphoblastic leukaemia (CCCG-ALL-2015): an open-label, multicentre, randomised, phase 3, non-inferiority trial. Lancet Oncol 22 (9): 1322-1332, 2021.
- Guolla L, Breitbart S, Foroutan F, et al.: Impact of vincristine-steroid pulses during maintenance for B-cell pediatric ALL: a systematic review and meta-analysis. Blood 141 (24): 2944-2954, 2023.
- Strauss AJ, Su JT, Dalton VM, et al.: Bony morbidity in children treated for acute lymphoblastic leukemia. J Clin Oncol 19 (12): 3066-72, 2001.
- Warris LT, van den Heuvel-Eibrink MM, Aarsen FK, et al.: Hydrocortisone as an Intervention for Dexamethasone-Induced Adverse Effects in Pediatric Patients With Acute Lymphoblastic Leukemia: Results of a Double-Blind, Randomized Controlled Trial. J Clin Oncol 34 (19): 2287-93, 2016.
- Hoppmann AL, Chen Y, Landier W, et al.: Individual prediction of nonadherence to oral mercaptopurine in children with acute lymphoblastic leukemia: Results from COG AALL03N1. Cancer 127 (20): 3832-3839, 2021.
- Bhatia S, Landier W, Hageman L, et al.: Systemic Exposure to Thiopurines and Risk of Relapse in Children With Acute Lymphoblastic Leukemia: A Children's Oncology Group Study. JAMA Oncol 1 (3): 287-95, 2015.
- Landier W, Chen Y, Hageman L, et al.: Comparison of self-report and electronic monitoring of 6MP intake in childhood ALL: a Children's Oncology Group study. Blood 129 (14): 1919-1926, 2017.
Terapia dirigida al sistema nervioso central para la leucemia linfoblástica aguda infantil
Aspectos generales de los regímenes terapéuticos dirigidos al sistema nervioso central
En el momento del diagnóstico, cerca del 3 % de los pacientes tienen enfermedad SNC3 (definida por una muestra de líquido cefalorraquídeo [LCR] con ≥5 glóbulos blancos [GB]/μl, linfoblastos o parálisis de nervios craneales). Sin embargo, a menos que se dirija una terapia específica al sistema nervioso central (SNC), la mayoría de los niños presentará con el tiempo leucemia en el SNC evidente sin importar si se detectaron linfoblastos o no en el LCR en el momento del diagnóstico inicial. Por consiguiente, todos los niños con leucemia linfoblástica aguda (LLA) deben recibir una quimioterapia combinada sistémica y alguna forma de profilaxis en el SNC.
Debido a que el SNC es un sitio santuario (es decir, un espacio anatómico de poca penetración para muchos de los fármacos quimioterapéuticos que a menudo se usan para el tratamiento de la LLA), las terapias específicas dirigidas al SNC se deben iniciar rápido para eliminar la enfermedad en el SNC sintomática en el momento del diagnóstico y para prevenir la recaída en el SNC en todos los pacientes. Tradicionalmente, las tasas de supervivencia de los niños con LLA mejoraron mucho luego de que se empezaran a añadir terapias dirigidas al SNC a los regímenes de tratamiento.
Las opciones de tratamiento estándar para la terapia dirigida al sistema nervioso central son las siguientes:
- Quimioterapia intratecal.
- Quimioterapia sistémica dirigida al SNC.
- Radioterapia craneal.
Todas estas modalidades terapéuticas tienen una función en el tratamiento y la prevención de la leucemia en el SNC. El uso de una combinación de quimioterapia intratecal y quimioterapia sistémica dirigida al SNC es estándar. La radiación craneal se reserva para situaciones especiales.
El tipo de terapia dirigida al SNC se basa en el riesgo de recaída en el SNC; los pacientes de riesgo más alto reciben tratamientos más intensivos. Los datos indican que los siguientes grupos de pacientes tienen un aumento de riesgo de recaída en el SNC:
- Pacientes con 5 GB/µl o más, y blastocitos en el LCR (SNC3) en el momento del diagnóstico.
- Los pacientes con blastocitos en el LCR, pero menos de 5 GB/µl (SNC2) quizás también tengan un riesgo alto de recaída en el SNC, sin embargo, este riesgo se elimina casi por completo cuando reciben más dosis de quimioterapia intratecal, en especial, durante la fase de inducción.
- Pacientes con LLA-T, en especial, los que tienen recuentos leucocitarios altos en la sangre periférica durante la presentación inicial.
- Los pacientes con una punción lumbar traumática en la que se observan blastocitos en el momento del diagnóstico quizás tengan un riesgo alto de recaída en el SNC. En algunos protocolos de tratamiento, estos pacientes reciben terapia dirigida al SNC más intensiva.
Los regímenes terapéuticos dirigidos al SNC para la LLA infantil recién diagnosticada se presentan en el Cuadro 11.
| Estado de la enfermedad | Opciones de tratamiento estándar | |
|---|---|---|
| LLA = leucemia linfoblástica aguda; SNC = sistema nervioso central; SNC3 = líquido cefalorraquídeo con 5 o más glóbulos blancos/µl, resultado positivo para blastocitos en la prueba con citocentrífuga o parálisis de nervios craneales. | ||
| aEl fármaco solo no entra en el SNC, pero produce el agotamiento de la asparagina en el líquido cefalorraquídeo. | ||
| Leucemia linfoblástica aguda de riesgo estándar | Quimioterapia intratecal | |
| Metotrexato solo | ||
| Metotrexato con citarabina e hidrocortisona | ||
| Quimioterapia sistémica dirigida al SNC | ||
| Dexametasona | ||
| L-asparaginasaa | ||
| Dosis altas de metotrexato con rescate de leucovorina | ||
| Dosis escalonadas de metotrexato intravenoso (sin rescate de leucovorina) | ||
| Leucemia linfoblástica aguda de riesgo alto y riesgo muy alto | Quimioterapia intratecal | |
| Metotrexato solo | ||
| Metotrexato con citarabina e hidrocortisona | ||
| Quimioterapia sistémica dirigida al SNC | ||
| Dexametasona | ||
| L-asparaginasaa | ||
| Dosis altas de metotrexato con rescate de leucovorina | ||
| Radioterapia craneal | ||
Un objetivo principal de los ensayos clínicos actuales de LLA es administrar una terapia dirigida al SNC eficaz y, al mismo tiempo, reducir al mínimo los efectos tóxicos neurológicos y otros efectos tardíos.
Quimioterapia intratecal
Todos los regímenes terapéuticos para la LLA infantil incluyen quimioterapia intratecal. La quimioterapia intratecal se suele iniciar al principio de la inducción, se intensifica durante la consolidación y, en muchos protocolos, continúa durante la fase de mantenimiento.
La quimioterapia intratecal suele incluir una de las siguientes opciones:
- Metotrexato solo.
- Metotrexato con citarabina e hidrocortisona (quimioterapia intratecal triple).
A diferencia de la citarabina intratecal, el metotrexato intratecal tiene un efecto sistémico significativo que quizás contribuya a prevenir la recaída en la médula.
Quimioterapia sistémica dirigida al sistema nervioso central
Además de las terapias dirigidas directamente al encéfalo y el líquido cefalorraquídeo, los fármacos de administración sistémica también son un componente eficaz para la profilaxis en el SNC. Los siguientes fármacos de administración sistémica proporcionan algún grado de profilaxis en el SNC:
- Dexametasona.
- L-asparaginasa (no penetra en el LCR, pero produce disminución de la asparagina en el LCR).
- Dosis altas de metotrexato con rescate de leucovorina.
- Dosis intravenosas (IV) escalonadas de metotrexato sin rescate de leucovorina.
Evidencia (quimioterapia sistémica dirigida al sistema nervioso central):
- En un estudio aleatorizado del Children's Cancer Group (CCG) con pacientes de riesgo estándar que recibieron el mismo programa de dosis de metotrexato intratecal sin irradiación craneal, se encontró que la dexametasona oral se relacionó con una disminución del 50 % en la tasa de recaída en el SNC en comparación con la prednisona oral.
- En otro ensayo sobre LLA de riesgo estándar (COG-1991), el uso de dosis escalonadas de metotrexato IV sin rescate de leucovorina redujo de forma significativa la recaída en el SNC en comparación con las dosis bajas de metotrexato oral estándar administradas durante cada una de las 2 fases de mantenimiento provisional.
- En un ensayo clínico aleatorizado dirigido por el antiguo Pediatric Oncology Group, los pacientes con LLA-T que recibieron dosis altas de metotrexato tuvieron una tasa de recaída en el SNC significativamente inferior que quienes no recibieron dosis altas de este medicamento.
Radioterapia craneal
La proporción de pacientes que reciben radioterapia craneal ha disminuido de manera significativa con el tiempo. Ahora la mayoría de niños con LLA recién diagnosticada no reciben radioterapia craneal durante el tratamiento. Muchos grupos administran radioterapia craneal solo a los pacientes con el riesgo más alto de recaída en el SNC, como los pacientes con leucemia en el SNC documentada en el momento del diagnóstico (como se definió antes) (≥5 GB/μl y blastocitos; SNC3), o fenotipo de células T con recuento de GB alto en el momento de la presentación inicial. Algunos centros no usan la irradiación craneal para ningún paciente.
En los pacientes que reciben radioterapia, la dosis de radioterapia craneal se ha disminuido de manera significativa y la administración de irradiación medular no es estándar.
En los ensayos en curso se busca determinar si es posible eliminar la radioterapia del tratamiento de todos los niños con LLA recién diagnosticada sin que esto afecte la supervivencia ni aumente la tasa de toxicidad de las terapias iniciales y de rescate. En un metanálisis de ensayos aleatorizados de terapias dirigidas al SNC se confirmó que la quimioterapia intratecal puede reemplazar la radioterapia en la mayoría de los pacientes con LLA recién diagnosticada. Es posible que se necesite tratamiento sistémico adicional según los fármacos y la intensidad del tratamiento usado.; [Nivel de evidencia B1]
Terapia dirigida al sistema nervioso central para los pacientes de riesgo estándar
La quimioterapia intratecal sin radioterapia craneal en el contexto de una quimioterapia sistémica adecuada produce tasas de recaída en el SNC inferiores al 5 % en los niños con LLA de riesgo estándar.
El uso de radioterapia craneal no es un componente necesario de la terapia dirigida al SNC para estos pacientes. En algunos regímenes se usa quimioterapia intratecal triple (metotrexato, citarabina e hidrocortisona), mientras que en otros se usa metotrexato intratecal solo durante todo el tratamiento.
Evidencia (quimioterapia intratecal triple vs. metotrexato intratecal):
- En el estudio CCG-1952 con pacientes de riesgo estándar del Instituto Nacional del Cáncer (NCI) se comparó la eficacia relativa y la toxicidad de la quimioterapia intratecal triple (metotrexato, citarabina e hidrocortisona) con el metotrexato como monoterapia intratecal en pacientes que no recibieron radiación.
- No hubo diferencias significativas en los efectos tóxicos en el SNC o fuera de este.
- Aunque la quimioterapia intratecal triple se relacionó con una tasa más baja de recaída aislada en el SNC (3,4 % ± 1,0 %, en comparación con 5,9 % ± 1,2 % para el metotrexato intratecal; P = 0,004), no hubo ninguna diferencia en las tasas de supervivencia sin complicaciones (SSC).
- La reducción de la tasa de recaída en el SNC fue importante, en particular en los pacientes con estado SNC2 en el momento del diagnóstico (linfoblastos observados en la citocentrifugación del LCR, pero con <5 GB/campo de gran aumento en el recuento de células del LCR). La tasa de recaída aislada en el SNC fue del 7,7 % (± 5,3 %) en los pacientes SNC2 que recibieron quimioterapia intratecal triple, en comparación con el 23,0 % (± 9.5%) en aquellos que recibieron metotrexato intratecal solo (P = 0,04).
- Hubo más recaídas en la médula ósea en el grupo que recibió quimioterapia intratecal triple, lo que produjo una tasa de supervivencia general (SG) más precaria (90,3 % ± 1,5 %) en comparación con el grupo de metotrexato intratecal (94,4 % ± 1,1 %; P = 0,01).
- Cuando el análisis se limitó a los pacientes con LLA-B y respuesta temprana rápida (médula de tipo M1 en el día 14), no hubo ninguna diferencia entre la quimioterapia intratecal triple y la quimioterapia intratecal única en términos de tasas de recaída en el SNC, o tasas de SG o SSC.
- Es necesario interpretar los hallazgos de este ensayo en el contexto de las otras terapias que reciben estos pacientes. En otros ensayos, la dexametasona se ha relacionado con tasas de recaída en el SNC más bajas y mejores tasas de SSC en los pacientes de riesgo estándar, pero este medicamento no se usó en el ensayo CCG-1952 (la prednisona fue el único corticoesteroide usado). No está claro si los resultados del ensayo CCG-1952 se pueden generalizar a los protocolos que incluyen el uso de dexametasona y otras terapias sistémicas dirigidas al SNC.
- En un estudio de seguimiento del funcionamiento neurocognitivo en los 2 grupos, no se encontraron diferencias significativas desde el punto de vista clínico.[Nivel de evidencia A3]
Terapia dirigida al sistema nervioso central para pacientes de riesgo alto y de riesgo muy alto sin compromiso del sistema nervioso central
Quimioterapia intratecal
Los abordajes de terapia intratecal también se han estudiado en pacientes de riesgo alto.
Evidencia (quimioterapia intratecal triple vs. metotrexato intratecal):
- En el estudio del COG AALL1131 (NCT02883049) para pacientes con LLA-B de riesgo alto del NCI y pacientes de riesgo estándar del NCI con respuesta temprana lenta (definida como ERM en sangre periférica el día 8 o ERM en la médula el día 29), los participantes de 1 a 30 años se asignaron al azar para recibir metotrexato intratecal posinducción o quimioterapia intratecal triple (metotrexato, citarabina e hidrocortisona). Los pacientes con enfermedad SNC3 no fueron aptos para participar, y los pacientes de este ensayo no recibieron radiación craneal. Se administraron un total de 21 a 26 dosis de terapia intratecal posinducción. Durante la evaluación inicial, se realizaron pruebas neurocognitivas a un subgrupo de pacientes de entre 6 y 12 años.
- Las tasas de supervivencia sin enfermedad (SSE) a 5 años fueron del 93,2 % (± 2,1 %) en los pacientes asignados al azar para recibir metotrexato intratecal y del 90,6 % (± 2,3 %) (P = 0,85) en los pacientes asignados para recibir quimioterapia intratecal triple.
- Las tasas de SG fueron del 96,3 % (± 1,5 %) en los pacientes que recibieron metotrexato intratecal y del 96,7 % (± 1,4 %) (P = 0,77) en los pacientes que recibieron quimioterapia intratecal triple.
- No hubo diferencias entre los 2 grupos en la incidencia acumulada de recaída aislada en la médula ósea, recaída aislada en el SNC, ni en la combinación de recaídas en la médula ósea y el SNC.
- Tampoco hubo diferencias significativas en los efectos tóxicos neurológicos ni en las pruebas de funcionamiento neurocognitivo durante el tratamiento de los pacientes que recibieron metotrexato intratecal, en comparación con los que recibieron quimioterapia intratecal triple.
Radioterapia craneal
Hay controversia sobre el uso de radioterapia craneal en los pacientes de riesgo alto y riesgo muy alto, aunque cada vez hay mayor consenso en cuanto a que la mayoría de los pacientes no necesitan radioterapia craneal. Las indicaciones para el uso de radioterapia craneal en algunos regímenes terapéuticos son las siguientes:
- Pacientes con fenotipo de células T y recuento de GB inicial alto.
- Pacientes con LLA-B de riesgo alto y recuentos leucocitarios muy altos en el momento de la presentación inicial o anomalías citogenéticas adversas.
En las últimas dos décadas ha disminuido la proporción de pacientes que reciben radioterapia y la dosis de radioterapia que se administra.
Evidencia (radioterapia craneal):
- En un ensayo realizado entre 1990 y 1995, el grupo Berlin-Frankfurt-Münster (BFM) demostró que una dosis reducida de radiación profiláctica (12 Gy en lugar de 18 Gy) ofrecía una profilaxis eficaz en el SNC de los pacientes en riesgo alto.
- En el ensayo de seguimiento realizado por el grupo BFM entre 1995 y 2000 (BFM-95), se administró radioterapia craneal a cerca del 20 % de los pacientes (en comparación con el 70 % en el ensayo anterior), incluidos los pacientes con fenotipo de células T, respuesta temprana lenta (según la medición del recuento de blastocitos en sangre periférica después de una semana de la profase con corticoesteroides) o anomalías citogenéticas adversas.
- A pesar de que la tasa de recaídas aisladas en el SNC fue más alta en los pacientes de riesgo más alto que no recibieron radiación, comparados con las cohortes históricas (pacientes irradiados), la tasa general de SSC no fue significativamente diferente.
- Varios grupos, como el St. Jude Children's Research Hospital (SJCRH), el Dutch Childhood Oncology Group (DCOG) y la European Organization for Research and Treatment of Cancer (EORTC), publicaron resultados de ensayos en los que se omitió la radioterapia craneal en todos los pacientes, incluso en los subconjuntos de riesgo alto. En la mayoría de estos ensayos se incluyeron por lo menos 4 dosis altas de metotrexato durante la consolidación de posinducción y un aumento en la frecuencia de la quimioterapia intratecal. En los estudios del SJCRH y DCOG también se incluyeron dosis altas repetidas de vincristina y dexametasona y dosis intensificadas de pegaspargasa, mientras que en los ensayos de la EORTC se incluyeron dosis altas de metotrexato adicionales y múltiples dosis altas de citarabina durante las fases de tratamiento de posinducción para los pacientes con un estado SNC3 (LCR con ≥5 GB/µl y resultado positivo para blastocitos en la prueba con citocentrífuga).
- La incidencia acumulada a 5 años de recaída aislada en el SNC en estos ensayos osciló entre el 2 % y el 4 %, aunque algunos subconjuntos de pacientes tuvieron una tasa significativamente más alta de recaída en el SNC. En el estudio del SJCRH, las características clínicas relacionadas con un riesgo significativamente más alto de recaída aislada en el SNC incluyeron el fenotipo de células T, la translocación t(1;19) o la presencia de blastocitos en el LCR en el momento del diagnóstico.
- La tasa de SSC general fue del 85,6 % en el estudio del SJCRH y del 81 % en el estudio del DCOG; ambos coincidieron con los resultados de ensayos clínicos contemporáneos en los que algunos pacientes recibieron radioterapia profiláctica, pero esta tasa fue más baja en el ensayo de la EORTC (SSC a 8 años del 69,6 %).
- En el estudio del SJCRH, 33 de 498 pacientes (6,6 %) en primera remisión con características de riesgo alto (26 pacientes con enfermedad residual mínima [ERM] alta, 6 con LLA positiva para BCR::ABL1 y 1 paciente casi haploide) recibieron un trasplante de células madre hematopoyéticas alogénico, que incluyó irradiación corporal total.
- En un metanálisis de datos combinados de más de 16 000 pacientes tratados entre 1996 y 2007 por 10 grupos cooperativos, el uso de radioterapia craneal no afectó la SG a 5 años ni la incidencia acumulada de ningún evento adverso.
- En análisis de subgrupos de subpoblaciones de pacientes de riesgo alto, solo los que tenían un estado SNC3 en el momento del diagnóstico se beneficiaron de la radioterapia craneal; se observó una tasa significativamente inferior de recaídas en el SNC (aislada o de cualquier tipo) en los pacientes irradiados. Sin embargo, incluso dentro de este subgrupo, las tasas de SG fueron similares con el uso de radioterapia o sin este.
- Este estudio indica que la radioterapia craneal puede no ser un componente esencial del tratamiento, incluso en los pacientes de riesgo alto. Sin embargo, la interpretación se complica debido a la gran variedad de tratamientos administrados a los pacientes por los diferentes grupos cooperativos.
- En el ensayo EORTC-58832 que se realizó entre 1983 y 1989 se incluyeron pacientes con LLA de riesgo medio y riesgo alto. Los pacientes se asignaron al azar a recibir radiación craneal después de la intensificación y antes de la terapia de mantenimiento, o a no recibir radiación craneal.[Nivel de evidencia A1]
- Las tasas de SSC y SG a 25 años fueron similares en los 2 grupos del ensayo: la tasa de SSC fue del 59,5 % y la tasa de SG fue del 78,1 % en los pacientes que no recibieron radiación craneal; la tasa de SSC fue del 60,5 % y la tasa de SG fue del 78,5 % en los pacientes que recibieron radiación craneal.
- Hubo una reducción intensa de la tasa de efectos adversos tardíos en el SNC en los pacientes que no recibieron radioterapia craneal.
- La incidencia de segundas neoplasias también disminuyó en los pacientes que no recibieron radiación (7,3 %) en comparación con aquellos que sí recibieron radiación (13 %) (CRI, 0,43). Los meningiomas explicaron el aumento de la incidencia de segundas neoplasias en el grupo de radiación craneal.
Terapia dirigida al sistema nervioso central en los pacientes con enfermedad SNC3 en el momento del diagnóstico
El tratamiento de los pacientes con LLA y enfermedad en el SNC evidente del punto de vista clínico (≥5 GB/CGA y blastocitos en la prueba con citocentrífuga; SNC3 con parálisis de nervios craneales) en el momento del diagnóstico suele incluir quimioterapia intratecal y radioterapia craneal (dosis habitual de 18 Gy). Ya no se usa la radiación raquídea.
Evidencia (radioterapia craneal):
- El SJCRH, el DCOG y la EORTC publicaron los resultados de ensayos en los que se omitió el uso de radioterapia craneal en todos los pacientes, incluso en los subconjuntos de riesgo alto. En la mayoría de estos ensayos se usaron por lo menos 4 dosis altas de metotrexato durante la consolidación de posinducción y un aumento de la frecuencia de la quimioterapia intratecal. En el estudio del SJCRH también se usaron dosis acumuladas más altas de antraciclinas, en comparación con los ensayos del Children’s Oncology Group (COG), además de dosis altas repetidas de vincristina y dexametasona e intensificación de la dosis de pegaspargasa, mientras que, en los ensayos del SJCRH, se usaron dosis altas de metotrexato adicionales y múltiples dosis altas de citarabina durante las fases de posinducción del tratamiento para los pacientes con estado SCN3 (LCR ≥ 5 GB/µl y resultado positivo para blastocitos en la prueba con citocentrífuga).
- En el estudio del SJCRH Total XV (TOTXV), los pacientes con estado SNC3 (N = 9) se trataron sin radioterapia craneal (tasa de SSC a 5 años observada del 43 % ± 23 %; tasa de SG del 71 % ± 22 %). En este estudio, la leucemia en el SNC en el momento del diagnóstico (definida como estado SNC3 o punción lumbar traumática con blastocitos) fue un factor pronóstico independiente de una SSC inferior.
- En el ensayo DCOG-9, la tasa de SSC a 5 años de los pacientes con SNC3 (n = 21) tratados sin radioterapia craneal fue del 67 % (± 10 %).
- En el ensayo de EORTC, la tasa de SSC a 8 años de los pacientes con SNC3 (n = 49) tratados sin radioterapia craneal fue del 68 %. La incidencia acumulada de recaída aislada en el SNC de estos pacientes fue del 9,4 %.[Nivel de evidencia B4]
- En un metanálisis de datos agrupados de más de 16 000 pacientes tratados entre 1996 y 2007 por 10 grupos cooperativos, se evaluó si el uso de radioterapia craneal afectó el desenlace en el subgrupo de pacientes de riesgo alto.
- En análisis de subgrupos de subpoblaciones de pacientes de riesgo alto, solo los que tenían un estado SNC3 en el momento del diagnóstico se beneficiaron de la radioterapia craneal; se observó una tasa significativamente inferior de recaídas en el SNC (aislada o de cualquier tipo) en los pacientes irradiados. Sin embargo, incluso dentro de este subgrupo, las tasas de SG fueron similares con el uso de radioterapia o sin este.
Se necesitan estudios prospectivos más grandes para determinar por completo la inocuidad de la omisión de la radioterapia craneal para los pacientes con SNC3.
Opciones de terapia dirigida al sistema nervioso central en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el NCI se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
Toxicidad de la terapia dirigida al sistema nervioso central
Los efectos tóxicos de la terapia dirigida al SNC para la LLA infantil pueden ser agudos, subagudos y tardíos. Para obtener más información, consultar la sección Efectos tardíos en el sistema nervioso central en Efectos tardíos del tratamiento anticanceroso en la niñez.
Efectos tóxicos agudos y subagudos
Las convulsiones son el efecto secundario agudo más común de la quimioterapia intratecal sola. Hasta un 5 % de los pacientes con LLA no irradiados que reciben dosis frecuentes de quimioterapia intratecal presentarán al menos una convulsión durante el tratamiento. Se observaron tasas más altas de convulsiones con el uso de regímenes de consolidación que incluyeron 12 cursos de dosis intermedias de metotrexato IV (1 g/m2) cada 2 semanas con quimioterapia intratecal. El uso de metotrexato por vía intratecal o en dosis altas IV también se relaciona con un síndrome similar a un accidente cerebrovascular que, en la mayoría de los casos, es transitorio.
Los pacientes de LLA que presentan convulsiones durante el tratamiento y que reciben anticonvulsivos no deben recibir fenobarbital ni fenitoína, porque estos medicamentos a veces aumentan la depuración de algunos fármacos quimioterapéuticos y afectan de manera adversa el desenlace.
Efectos tóxicos tardíos
Los efectos tardíos relacionados con las terapias dirigidas al SNC incluyen neoplasias subsiguientes, trastornos neuroendocrinos, leucoencefalopatía y alteraciones neurocognitivas.
Las neoplasias subsiguientes se observan sobre todo en sobrevivientes que recibieron radioterapia craneal. Los meningiomas son las segundas neoplasias que se observan con mayor frecuencia y en general son de bajo potencial maligno. Sin embargo, es posible que también se presenten lesiones de grado alto. En un estudio retrospectivo del SJCRH con más de 1290 pacientes de LLA que nunca recayeron, la incidencia acumulada a 30 años de una neoplasia subsiguiente en el SNC fue del 3 %. Y al descontar los meningiomas, la incidencia acumulada a 30 años fue del 1,17 %. Casi todas estas neoplasias subsiguientes en el SNC se presentaron en pacientes previamente irradiados.
Las alteraciones neurocognitivas, que varían en gravedad y consecuencias funcionales, se han documentado en sobrevivientes a largo plazo de LLA tratados con radioterapia o sin esta. En general, los pacientes que reciben quimioterapia intratecal sin radioterapia craneal presentan secuelas neurocognitivas menos graves que los pacientes irradiados; el deterioro estriba en disminuciones relativamente moderadas en un número restringido de dominios del funcionamiento neuropsicológico. En los pacientes que reciben radioterapia craneal, la frecuencia y gravedad de los efectos tóxicos dependen de la dosis. Los pacientes tratados con 18 Gy de radioterapia craneal tienen un riesgo más bajo de alteraciones graves que los pacientes tratados con dosis de 24 Gy o superiores. En muchos estudios, se notificó que una edad más baja en el momento del diagnóstico y el sexo femenino se relacionan con un riesgo más alto de efectos neurocognitivos tardíos.
En varios estudios también se evaluó el efecto de otros componentes del tratamiento en la presentación de alteraciones neurocognitivas tardías. No se observó ninguna diferencia de importancia clínica en una comparación de los resultados neurocognitivos de los pacientes tratados con metotrexato versus quimioterapia intratecal triple.[Nivel de evidencia C1] Hay controversia sobre el supuesto aumento del riesgo de alteraciones neurocognitivas en los pacientes que recibieron dexametasona. En un estudio del SJCRH con sobrevivientes a largo plazo no irradiados, el tratamiento con dexametasona se relacionó con aumento del riesgo de alteraciones de la atención y el funcionamiento ejecutivo. Por el contrario, en pruebas neurocognitivas a largo plazo de 92 niños con antecedentes de LLA de riesgo estándar que recibieron dexametasona o prednisona durante el tratamiento, no se demostraron diferencias significativas en el funcionamiento cognitivo según la aleatorización al grupo de corticoesteroides.
Evidencia (efectos tardíos neurocognitivos de la radiación craneal):
- En un estudio del SJCRH, 567 adultos sobrevivientes a largo plazo de LLA infantil se sometieron a pruebas neurocognitivas (tiempo medio desde el diagnóstico, 26 años).
- Los pacientes tratados con 24 Gy de radioterapia craneal exhibieron las tasas más altas de deterioro. Hasta un tercio de estos pacientes presentaron deterioro (definido como puntajes de 2 o más desviaciones estándar por debajo de las normas nacionales ajustadas por edad) de la atención, la memoria, la velocidad de procesamiento y el funcionamiento ejecutivo.
- Un número significativamente más bajo de pacientes que habían recibido 18 Gy de radioterapia craneal exhibieron deterioro grave, en comparación con los que recibieron 24 Gy. En general, no hubo diferencias significativas en las tasas de deterioro entre los sobrevivientes no irradiados y quienes recibieron 18 Gy de radioterapia craneal. Sin embargo, el grupo que recibió 18 Gy tuvo un aumento del riesgo de dificultades académicas.
- Además de la relación con la dosis, el efecto neurocognitivo de la radioterapia craneal también dependió de la edad en el momento del diagnóstico; el deterioro fue más frecuente en pacientes diagnosticados a una edad menor.
- En un estudio se comparó el deterioro en la memoria de los pacientes que recibieron 18 Gy de radioterapia craneal (n = 127) versus 24 Gy de radioterapia craneal (n = 138).
- Los sobrevivientes a largo plazo que recibieron 24 Gy de radioterapia craneal presentaron deterioros significativos en la memoria inmediata y tardía, en comparación con los sobrevivientes que recibieron 18 Gy.
- En un ensayo aleatorizado en el que se compararon pacientes con LLA de riesgo estándar que recibieron irradiación (dosis de 18 Gy) y pacientes que no recibieron irradiación, se observaron los siguientes resultados:[Nivel de evidencia A3]
- El funcionamiento cognitivo en ambos grupos (evaluado en una mediana de 6 años después del diagnóstico) se ubicó en el intervalo promedio, y solo se observaron diferencias sutiles en las aptitudes cognitivas entre los grupos.
- En un ensayo aleatorizado, la radioterapia hiperfraccionada (en una dosis de 18 Gy) no redujo los efectos neurológicos tardíos, en comparación con la radioterapia fraccionada convencional. El funcionamiento cognitivo en ambos grupos no se deterioró de forma significativa.
Evidencia (efectos tardíos neurocognitivos en pacientes no irradiados):
- En el estudio del SJCRH de seguimiento a largo plazo de 567 adultos sobrevivientes a largo plazo, algunos pacientes no irradiados también tuvieron deterioro neurocognitivo.
- Los puntajes medios de las pruebas ajustados por edad en los pacientes no irradiados fueron muy similares a las normas nacionales previstas. Sin embargo, cerca del 15 % de los sobrevivientes no irradiados que participaron en este estudio demostraron deterioro en algún dominio, como atención, memoria, velocidad de procesamiento y funcionamiento ejecutivo.
- Pese al deterioro en las pruebas neurocognitivas, en general, los logros académicos y la situación laboral de los sobrevivientes de LLA evaluados fueron similares a las proporciones esperadas ajustadas por edad y sexo, según los datos del censo de población de los Estados Unidos.
- En otro estudio del SJCRH, los pacientes que participaron en el estudio Total Study XV (en el que se omitió la radioterapia craneal en todos los pacientes) se sometieron a evaluaciones neuropsicológicas completas en el momento de la inducción, el final del mantenimiento y 2 años después de terminar el tratamiento.
- El funcionamiento neurocognitivo fue en gran medida apropiado para la edad 2 años después de terminar el tratamiento, sin indicios de exceso de deterioro en las mediciones de funcionamiento intelectual, aptitudes académicas, aprendizaje y memoria. Los problemas para mantener la atención fueron más frecuentes en esta población, en comparación con los problemas esperados (expectativas normativas).
- Los pacientes de riesgo alto que recibieron quimioterapia más intensiva dirigida al SNC (incluso dosis altas de metotrexato y más dosis de quimioterapia intratecal) se expusieron a mayor riesgo de problemas de atención, velocidad de procesamiento y rendimiento académico.
- En un estudio posterior del SJCRH, 400 pacientes del estudio Total XVI (inscritos entre 2007 y 2017) se sometieron a pruebas neurocognitivas después de completar el tratamiento (cerca de 3 años después del diagnóstico). Ninguno de los pacientes recibió radiación craneal, pero el número de dosis de quimioterapia intratecal triple varió según el grupo de riesgo y el estado del SNC. A los pacientes con LLA de riesgo bajo se les recetaron entre 13 y 21 dosis, mientras que a los pacientes con LLA de riesgo estándar o alto se les recetaron 16 a 27 dosis.
- En comparación con las expectativas relativas a la edad, la cohorte completa de pacientes presentó alteraciones cognitivas en varios dominios, como atención, memoria operativa, funcionamiento ejecutivo, motricidad fina y habilidades de adaptación.
- En el análisis de todos los pacientes con LLA de riesgo bajo no se encontró correlación entre el número de dosis de quimioterapia intratecal triple (<20 vs. >21) y los resultados neurocognitivos.
- En los pacientes con LLA de riesgo estándar o alto, quienes recibieron el número de dosis más alto de quimioterapia intratecal triple (>27) presentaron aumento del riesgo de alteración en comparación con quienes recibieron menos dosis, en especial, en la memoria operativa, la atención y la motricidad fina.
- Los pacientes con seguro público o quienes no contaban con seguro médico presentaron peores desenlaces neurocognitivos en comparación con los pacientes con seguro privado.
- Entre los pacientes con LLA de riesgo estándar y alto, los niños varones presentaron mayor riesgo de alteraciones neurocognitivas que las niñas, al igual que los pacientes más jóvenes en el momento del diagnóstico.
- En este estudio no se evaluó la presencia de manifestaciones clínicas de las alteraciones neurocognitivas identificadas por pruebas neurocognitivas al final del tratamiento ni su trayectoria en el tiempo.
- En el estudio del COG AALL06N1 (NCT00437060), los pacientes con LLA-B de riesgo alto que se habían inscrito en el ensayo AALL0232 se sometieron a una evaluación neurocognitiva. Estos pacientes habían sido asignados al azar para recibir dosis altas de metotrexato con rescate de leucovorina o metotrexato IV en dosis de aumento escalonado con pegasparaginasa durante la fase de mantenimiento provisional del tratamiento en el ensayo AALL0232. Después de completar el tratamiento, se llevó a cabo una evaluación neurocognitiva, que incluyó una evaluación del funcionamiento intelectual (coeficiente intelectual [IQ] estimado), la memoria de trabajo y la velocidad de procesamiento.
- La forma de administración del metotrexato no se relacionó con diferencias en los desenlaces neurocognitivos después de que se ajustara por etnia, raza, edad, sexo, tipo de seguro médico y tiempo sin tratamiento.
- Los sobrevivientes que tenían menos de 10 años en el momento del diagnóstico presentaron puntajes estimados de IQ y velocidad de procesamiento significativamente más bajos que los pacientes de edad más avanzada.
- Además, los pacientes con un seguro médico público presentaron puntajes estimados de IQ más bajos que los participantes con seguro médico privado o militar de Estados Unidos.
References
- Richards S, Pui CH, Gayon P, et al.: Systematic review and meta-analysis of randomized trials of central nervous system directed therapy for childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 60 (2): 185-95, 2013.
- Mahmoud HH, Rivera GK, Hancock ML, et al.: Low leukocyte counts with blast cells in cerebrospinal fluid of children with newly diagnosed acute lymphoblastic leukemia. N Engl J Med 329 (5): 314-9, 1993.
- Bürger B, Zimmermann M, Mann G, et al.: Diagnostic cerebrospinal fluid examination in children with acute lymphoblastic leukemia: significance of low leukocyte counts with blasts or traumatic lumbar puncture. J Clin Oncol 21 (2): 184-8, 2003.
- Gajjar A, Harrison PL, Sandlund JT, et al.: Traumatic lumbar puncture at diagnosis adversely affects outcome in childhood acute lymphoblastic leukemia. Blood 96 (10): 3381-4, 2000.
- Pullen J, Boyett J, Shuster J, et al.: Extended triple intrathecal chemotherapy trial for prevention of CNS relapse in good-risk and poor-risk patients with B-progenitor acute lymphoblastic leukemia: a Pediatric Oncology Group study. J Clin Oncol 11 (5): 839-49, 1993.
- Thyss A, Suciu S, Bertrand Y, et al.: Systemic effect of intrathecal methotrexate during the initial phase of treatment of childhood acute lymphoblastic leukemia. The European Organization for Research and Treatment of Cancer Children's Leukemia Cooperative Group. J Clin Oncol 15 (5): 1824-30, 1997.
- Rizzari C, Lanvers-Kaminsky C, Valsecchi MG, et al.: Asparagine levels in the cerebrospinal fluid of children with acute lymphoblastic leukemia treated with pegylated-asparaginase in the induction phase of the AIEOP-BFM ALL 2009 study. Haematologica 104 (9): 1812-1821, 2019.
- Bostrom BC, Sensel MR, Sather HN, et al.: Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: a report from the Children's Cancer Group. Blood 101 (10): 3809-17, 2003.
- Matloub Y, Bostrom BC, Hunger SP, et al.: Escalating intravenous methotrexate improves event-free survival in children with standard-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 118 (2): 243-51, 2011.
- Asselin BL, Devidas M, Wang C, et al.: Effectiveness of high-dose methotrexate in T-cell lymphoblastic leukemia and advanced-stage lymphoblastic lymphoma: a randomized study by the Children's Oncology Group (POG 9404). Blood 118 (4): 874-83, 2011.
- Pui CH, Howard SC: Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol 9 (3): 257-68, 2008.
- Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.
- Veerman AJ, Kamps WA, van den Berg H, et al.: Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004). Lancet Oncol 10 (10): 957-66, 2009.
- Vora A, Andreano A, Pui CH, et al.: Influence of Cranial Radiotherapy on Outcome in Children With Acute Lymphoblastic Leukemia Treated With Contemporary Therapy. J Clin Oncol 34 (9): 919-26, 2016.
- Pui CH, Sandlund JT, Pei D, et al.: Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children's Research Hospital. Blood 104 (9): 2690-6, 2004.
- Tubergen DG, Gilchrist GS, O'Brien RT, et al.: Prevention of CNS disease in intermediate-risk acute lymphoblastic leukemia: comparison of cranial radiation and intrathecal methotrexate and the importance of systemic therapy: a Childrens Cancer Group report. J Clin Oncol 11 (3): 520-6, 1993.
- Conter V, Aricò M, Valsecchi MG, et al.: Extended intrathecal methotrexate may replace cranial irradiation for prevention of CNS relapse in children with intermediate-risk acute lymphoblastic leukemia treated with Berlin-Frankfurt-Münster-based intensive chemotherapy. The Associazione Italiana di Ematologia ed Oncologia Pediatrica. J Clin Oncol 13 (10): 2497-502, 1995.
- Möricke A, Reiter A, Zimmermann M, et al.: Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 111 (9): 4477-89, 2008.
- Clarke M, Gaynon P, Hann I, et al.: CNS-directed therapy for childhood acute lymphoblastic leukemia: Childhood ALL Collaborative Group overview of 43 randomized trials. J Clin Oncol 21 (9): 1798-809, 2003.
- Moghrabi A, Levy DE, Asselin B, et al.: Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood 109 (3): 896-904, 2007.
- Matloub Y, Lindemulder S, Gaynon PS, et al.: Intrathecal triple therapy decreases central nervous system relapse but fails to improve event-free survival when compared with intrathecal methotrexate: results of the Children's Cancer Group (CCG) 1952 study for standard-risk acute lymphoblastic leukemia, reported by the Children's Oncology Group. Blood 108 (4): 1165-73, 2006.
- Mitchell CD, Richards SM, Kinsey SE, et al.: Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomized trial. Br J Haematol 129 (6): 734-45, 2005.
- Vrooman LM, Stevenson KE, Supko JG, et al.: Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol 31 (9): 1202-10, 2013.
- Kadan-Lottick NS, Brouwers P, Breiger D, et al.: Comparison of neurocognitive functioning in children previously randomly assigned to intrathecal methotrexate compared with triple intrathecal therapy for the treatment of childhood acute lymphoblastic leukemia. J Clin Oncol 27 (35): 5986-92, 2009.
- Salzer WL, Burke MJ, Devidas M, et al.: Impact of Intrathecal Triple Therapy Versus Intrathecal Methotrexate on Disease-Free Survival for High-Risk B-Lymphoblastic Leukemia: Children's Oncology Group Study AALL1131. J Clin Oncol 38 (23): 2628-2638, 2020.
- Schrappe M, Reiter A, Ludwig WD, et al.: Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 95 (11): 3310-22, 2000.
- Sirvent N, Suciu S, Rialland X, et al.: Prognostic significance of the initial cerebro-spinal fluid (CSF) involvement of children with acute lymphoblastic leukaemia (ALL) treated without cranial irradiation: results of European Organization for Research and Treatment of Cancer (EORTC) Children Leukemia Group study 58881. Eur J Cancer 47 (2): 239-47, 2011.
- Piette C, Suciu S, Bertrand Y, et al.: Long-term outcome evaluation of medium/high risk acute lymphoblastic leukaemia children treated with or without cranial radiotherapy in the EORTC 58832 randomized study. Br J Haematol 189 (2): 351-362, 2020.
- Mahoney DH, Shuster JJ, Nitschke R, et al.: Acute neurotoxicity in children with B-precursor acute lymphoid leukemia: an association with intermediate-dose intravenous methotrexate and intrathecal triple therapy--a Pediatric Oncology Group study. J Clin Oncol 16 (5): 1712-22, 1998.
- Bhojwani D, Sabin ND, Pei D, et al.: Methotrexate-induced neurotoxicity and leukoencephalopathy in childhood acute lymphoblastic leukemia. J Clin Oncol 32 (9): 949-59, 2014.
- Relling MV, Pui CH, Sandlund JT, et al.: Adverse effect of anticonvulsants on efficacy of chemotherapy for acute lymphoblastic leukaemia. Lancet 356 (9226): 285-90, 2000.
- Hijiya N, Hudson MM, Lensing S, et al.: Cumulative incidence of secondary neoplasms as a first event after childhood acute lymphoblastic leukemia. JAMA 297 (11): 1207-15, 2007.
- Waber DP, Turek J, Catania L, et al.: Neuropsychological outcomes from a randomized trial of triple intrathecal chemotherapy compared with 18 Gy cranial radiation as CNS treatment in acute lymphoblastic leukemia: findings from Dana-Farber Cancer Institute ALL Consortium Protocol 95-01. J Clin Oncol 25 (31): 4914-21, 2007.
- Jansen NC, Kingma A, Schuitema A, et al.: Neuropsychological outcome in chemotherapy-only-treated children with acute lymphoblastic leukemia. J Clin Oncol 26 (18): 3025-30, 2008.
- Espy KA, Moore IM, Kaufmann PM, et al.: Chemotherapeutic CNS prophylaxis and neuropsychologic change in children with acute lymphoblastic leukemia: a prospective study. J Pediatr Psychol 26 (1): 1-9, 2001 Jan-Feb.
- Copeland DR, Moore BD, Francis DJ, et al.: Neuropsychologic effects of chemotherapy on children with cancer: a longitudinal study. J Clin Oncol 14 (10): 2826-35, 1996.
- von der Weid N, Mosimann I, Hirt A, et al.: Intellectual outcome in children and adolescents with acute lymphoblastic leukaemia treated with chemotherapy alone: age- and sex-related differences. Eur J Cancer 39 (3): 359-65, 2003.
- Waber DP, Carpentieri SC, Klar N, et al.: Cognitive sequelae in children treated for acute lymphoblastic leukemia with dexamethasone or prednisone. J Pediatr Hematol Oncol 22 (3): 206-13, 2000 May-Jun.
- Krull KR, Brinkman TM, Li C, et al.: Neurocognitive outcomes decades after treatment for childhood acute lymphoblastic leukemia: a report from the St Jude lifetime cohort study. J Clin Oncol 31 (35): 4407-15, 2013.
- Kadan-Lottick NS, Brouwers P, Breiger D, et al.: A comparison of neurocognitive functioning in children previously randomized to dexamethasone or prednisone in the treatment of childhood acute lymphoblastic leukemia. Blood 114 (9): 1746-52, 2009.
- Armstrong GT, Reddick WE, Petersen RC, et al.: Evaluation of memory impairment in aging adult survivors of childhood acute lymphoblastic leukemia treated with cranial radiotherapy. J Natl Cancer Inst 105 (12): 899-907, 2013.
- Waber DP, Silverman LB, Catania L, et al.: Outcomes of a randomized trial of hyperfractionated cranial radiation therapy for treatment of high-risk acute lymphoblastic leukemia: therapeutic efficacy and neurotoxicity. J Clin Oncol 22 (13): 2701-7, 2004.
- Jacola LM, Krull KR, Pui CH, et al.: Longitudinal Assessment of Neurocognitive Outcomes in Survivors of Childhood Acute Lymphoblastic Leukemia Treated on a Contemporary Chemotherapy Protocol. J Clin Oncol 34 (11): 1239-47, 2016.
- Jacola LM, Conklin HM, Krull KR, et al.: The Impact of Intensified CNS-Directed Therapy on Neurocognitive Outcomes in Survivors of Childhood Acute Lymphoblastic Leukemia Treated Without Cranial Irradiation. J Clin Oncol 40 (36): 4218-4227, 2022.
- Hardy KK, Embry L, Kairalla JA, et al.: Neurocognitive Functioning of Children Treated for High-Risk B-Acute Lymphoblastic Leukemia Randomly Assigned to Different Methotrexate and Corticosteroid Treatment Strategies: A Report From the Children's Oncology Group. J Clin Oncol 35 (23): 2700-2707, 2017.
Tratamiento de posinducción para subgrupos específicos de leucemia linfoblástica aguda
T-cell childhood acute lymphoblastic leukemiaLeucemia linfoblástica aguda de células T
Tradicionalmente, los pacientes con leucemia linfoblástica aguda de células T (LLA-T) han tenido un pronóstico más precario que los niños con leucemia linfoblástica aguda de células B (LLA-B). En una revisión de un gran número de pacientes tratados en ensayos del Children's Oncology Group (COG) durante un periodo de 15 años, se encontró que el inmunofenotipo de células T continúa siendo un factor de pronóstico adverso en el análisis multivariante. Sin embargo, con el uso de los regímenes de tratamiento vigentes, los desenlaces de los niños con LLA-T se acercan a los de los niños con LLA-B. Por ejemplo, el Dana-Farber Cancer Institute (DFCI) ALL Consortium notificó una tasa de supervivencia sin complicaciones (SSC) a 5 años del 81 % y una tasa de supervivencia general (SG) del 90 % en los pacientes con LLA-T que se trataron en 2 ensayos clínicos consecutivos entre 2005 y 2015. Otro ejemplo es el ensayo del COG AALL0434 (NCT00408005) con pacientes de LLA-T, en el que se obtuvo una tasa de SSC a 5 años del 83,8 % y una tasa de SG del 89,5 %.
Opciones de tratamiento de la leucemia linfoblástica aguda de células T
Las opciones de tratamiento de la LLA-T son las siguientes:
- Quimioterapia con radioterapia craneal profiláctica o sin esta.
Evidencia (quimioterapia y radioterapia craneal profiláctica):
- En los protocolos del antiguo Pediatric Oncology Group (POG), el tratamiento de los niños con LLA-T era diferente al de los niños con LLA-B. En el ensayo POG-9404, los pacientes con LLA-T se trataron con el régimen del protocolo DFCI 87-001, que incluía múltiples dosis de L-asparaginasa y doxorrubicina durante la terapia de posinducción, así como radioterapia craneal profiláctica. Los pacientes se asignaron al azar al grupo de dosis altas de metotrexato o al grupo de control.
- Los pacientes asignados al azar para recibir dosis altas de metotrexato tuvieron desenlaces superiores (las tasas de SSC a 10 años fueron del 78 % con dosis altas de metotrexato vs. el 68 % en el grupo de control).
- En el estudio POG-9404, los pacientes se asignaron al azar para recibir doxorrubicina con dexrazoxano o sin este con el fin de determinar la eficacia del dexrazoxano para la prevención de la mortalidad tardía por problemas cardíacos.[Nivel de evidencia B1]
- No hubo diferencia en la SSC entre los pacientes con LLA-T que recibieron dexrazoxano y los pacientes que no recibieron dexrazoxano (dosis acumulada de doxorrubicina, 360 mg/m2).
- La frecuencia de efectos tóxicos de grados 3 y 4 durante el tratamiento fue similar entre los grupos aleatorizados, y no hubo diferencia en la incidencia acumulada de segundas neoplasias malignas. Después de 3 años del diagnóstico inicial, la fracción de acortamiento ventricular izquierda y el grosor de la pared ventricular izquierda fueron significativamente más precarios en los pacientes que recibieron doxorrubicina sola que en los pacientes que recibieron dexrazoxano, lo que indica que el dexrazoxano tuvo un efecto cardioprotector.
- En los datos combinados de 3 ensayos aleatorizados del COG de dexrazoxano comparado con doxorrubicina (P9404, P9425 y P9426) se encontró que al cabo de una mediana de seguimiento de 12,6 años, el dexrazoxano no afectó la supervivencia a largo plazo.[Nivel de evidencia A1]
- En los protocolos del antiguo Children’s Cancer Group (CCG), los niños con LLA-T recibieron el mismo tratamiento que los niños con LLA-B. La asignación al protocolo y al tratamiento se hizo según las características clínicas de los pacientes (por ejemplo, edad y recuento de glóbulos blancos [GB]) y la respuesta de la enfermedad a la terapia inicial. La mayoría de los niños con LLA-T cumple con los criterios de riesgo alto establecidos por el Instituto Nacional del Cáncer (NCI).
- En los resultados del ensayo CCG-1961 para la LLA de riesgo alto, que incluyó a pacientes con LLA-T, se indica que el uso de un régimen de Berlin-Frankfurt-Münster (BFM) intensificado con un solo curso de intensificación diferida produjo los mejores resultados para los pacientes con respuesta morfológica rápida a la terapia de inducción inicial (tasa de SSC estimada a 5 años, 83 %). Con este abordaje, los pacientes con recuentos de GB superiores o inferiores a 200 000 en el cuadro clínico inicial presentaron desenlaces similares.[Nivel de evidencia B1]
- Los resultados generales de los ensayos POG-9404 y CCG-1961 fueron similares, aunque en el POG-9404 se usaron dosis acumuladas más altas de antraciclinas y radioterapia craneal para todos los pacientes, mientras que en el CCG-1961 se usó radioterapia craneal solo para los pacientes con respuesta morfológica lenta.
- Entre los niños con LLA-T de riesgo estándar del NCI que se trataron en los ensayos CCG-1952, COG-1991 y POG-9404, las tasas de SSC a 7 años fueron similares a las de los pacientes tratados con los regímenes del CCG en los que se emplearon menos antraciclinas en una quimioterapia de base menos intensa, sin la irradiación craneal profiláctica del ensayo POG-9404. Sin embargo, los pacientes con LLA-T de riesgo estándar del NCI tuvieron SSC y SG inferiores en comparación con los pacientes con LLA-B de riesgo estándar del NCI que se trataron con los mismos regímenes en los ensayos CCG-1952 y COG-1991.
- En el ensayo del COG, los niños con LLA-T no se tratan usando los mismos protocolos que los niños con LLA-B.
- En estudios piloto del COG, se demostró la viabilidad de incorporar nelarabina (un análogo de nucleósido con acción confirmada en pacientes con enfermedad linfoblástica de células T en recaída y resistente al tratamiento) en el contexto de un régimen del BFM para los pacientes con LLA-T recién diagnosticada.
- En el estudio piloto se observó una tasa de SSC a 5 años del 73 % en todos los pacientes tratados con nelarabina y del 69 % en aquellos con una respuesta temprana lenta.
- En el ensayo del COG AALL0434 (NCT00408005) se incluyeron 1562 pacientes evaluables de 1 a 31 años con LLA-T. Los pacientes se sometieron a un régimen BFM intensificado y se asignaron al azar para recibir una fase de mantenimiento provisional con dosis altas de metotrexato y rescate de leucovorina, o metotrexato en dosis de aumento escalonado sin leucovorina, pero con pegaspargasa. Los pacientes de riesgo intermedio y riesgo alto también se asignaron al azar para recibir 6 ciclos de nelarabina durante el tratamiento de posinducción o para no recibir nelarabina. Casi todos los pacientes recibieron irradiación craneal profiláctica (12 Gy) o terapéutica (18 Gy). Solo el 10 % de los pacientes que se consideraron de riesgo bajo no se irradiaron. Los pacientes asignados al grupo de metotrexato en dosis de aumento escalonado recibieron radioterapia craneal antes que los pacientes asignados al grupo de dosis altas de metotrexato (semana 8 vs. 26). Los pacientes del grupo de metotrexato en dosis de aumento escalonado también recibieron 2 dosis adicionales de pegaspargasa. Los resultados fueron los siguientes:
- La tasa de SSC general a 5 años fue del 83,8 %, y la tasa de SG fue del 89,5 %.
- Los resultados indicaron una mejora de la supervivencia sin enfermedad (SSE) en los pacientes asignados al azar al grupo de metotrexato en dosis de aumento escalonado (tasa de SSE a 5 años, 91,5 %), en comparación con aquellos asignados al azar al grupo de dosis altas de metotrexato (tasa de SSC a 5 años, 85,3 %; P = 0,005).
- En los pacientes de riesgo intermedio y riesgo alto, el tratamiento con nelarabina se relacionó con un resultado superior (tasa de SSE a 5 años, 88,2 % con nelarabina vs. 82,1 % sin nelarabina; P = 0,029). La incidencia acumulada a 5 años de recaída en el sistema nervioso central (SNC) fue significativamente inferior en los pacientes tratados con nelarabina (1,3 vs 6,9 % en el grupo sin nelarabina).
- El mejor desenlace de los pacientes de riesgo intermedio y riesgo alto se observó en aquellos que se asignaron al azar a los grupos de metotrexato en dosis de aumento escalonado y de nelarabina (tasa de SSE a 5 años, 91,4 %). El peor desenlace se observó en los pacientes que se asignaron al azar para recibir metotrexato en dosis de aumento escalonado sin nelarabina (tasa de SSE a 5 años, 78,1 %).
- En los pacientes con enfermedad SNC3, todos los cuales se asignaron a recibir dosis altas de metotrexato y 18 Gy de radioterapia craneal, la nelarabina se relacionó con una SSE significativamente más alta.
- Los pacientes con fracaso de la inducción inicial (médula M3 en el día 29, n = 43) se asignaron de manera no aleatoria a recibir dosis altas de metotrexato y nelarabina; 20 de esos pacientes se discontinuaron del tratamiento del protocolo para someterse a un trasplante de células madre hematopoyéticas alogénico (TCMH) en la primera remisión completa (CR). La tasa general de SSC a 5 años para los pacientes con fracaso de la inducción fue del 53 %, sin diferencias entre los desenlaces del TCMH y la quimioterapia.
- En el ensayo sucesivo del COG AALL1231 (NCT02112916) en pacientes con LLA-T, los participantes se asignaron al azar a recibir un tratamiento con el inhibidor del proteasoma bortezomib o sin este durante las fases de inducción y de intensificación diferida. Los pacientes con fracaso de la inducción inicial (médula M3 en el día 29) o ERM alta (≥0,1 %) al final de la consolidación se asignaron de forma no aleatoria para recibir 3 bloques de quimioterapia intensificada adicionales después de la consolidación. Otros cambios al tratamiento que se hicieron de forma no aleatoria incluyeron el uso de dexametasona durante la inducción (en lugar de prednisona) y la omisión de radioterapia craneal en todos los pacientes, excepto en los de riesgo alto. Debido a que los resultados del ensayo AALL0434 no estaban disponibles en el momento en que comenzó el ensayo AALL1231, no se incluyó la nelarabina en la quimioterapia de base. El diseño original del estudio incluía 1200 pacientes, pero se interrumpió la inscripción de manera temprana (después de haberse inscrito 824 pacientes evaluables) cuando se publicaron los resultados de la aleatorización con nelarabina del ensayo AALL0434.
- Al comparar los pacientes con LLA-T asignados para recibir bortezomib versus los pacientes que no recibieron bortezomib, no hubo diferencias en la tasa de SSC a 4 años (82,9 vs. 81,5 %; P = 0,396) ni en la tasa de SG a 5 años (87,9 vs. 88,3 %; P = 0,469).
- Se hicieron análisis de subconjuntos donde se compararon pacientes similares con LLA-T que en el ensayo AALL0434 se asignaron para recibir 12 Gy de radioterapia craneal (90,8 % de los pacientes), pero que en el ensayo AALL1231 no recibieron radioterapia craneal (9,5 % de los pacientes). Si no se incluye a los pacientes que recibieron nelarabina (AALL0434) o bortezomib (AALL1231), las tasas de SSC a 4 años (P = 0,412), SG (P = 0,600), incidencia acumulada de recaída en el SNC (P = 0,456) y recaída general (P = 0,836) no presentaron diferencias significativas entre los estudios, lo que indica que la omisión de la radiación craneal (en el contexto de la quimioterapia de base del AALL1231) no afectó el desenlace de manera adversa.
- A pesar de que las tasas de SSC a 4 años fueron similares en ambos ensayos (P = 0,131), la tasa de SG del ensayo AALL1231 fue inferior a la del ensayo AALL0434 (87 vs. 90 %, P = 0,006).
- Si bien la incidencia acumulada de recaída fue similar en ambos estudios (P = 0,562), en el ensayo AALL1231 se observó una tasa más alta de mortalidad relacionada con el tratamiento, en comparación con la del ensayo AALL0434 (6,1 vs. 2,0 %). Las tasas de mortalidad relacionada con el tratamiento fueron más altas en el ensayo AALL1231 durante las fases de inducción (1,5 vs. 0,4 %, P = 0,002) y posinducción (3,9 vs. 2,1 %, P = 0,008) del tratamiento.
- A pesar de la intensificación de la quimioterapia de base, el desenlace de los pacientes con LLA-T asignados al grupo de riesgo muy alto (5,2 % de los pacientes) fue precario. La tasa de SSC a 4 años fue del 31,3 % en los pacientes del grupo sin bortezomib, en comparación con el 7,8 % en los pacientes del grupo con bortezomib (P = 0,033).
- En estudios piloto del COG, se demostró la viabilidad de incorporar nelarabina (un análogo de nucleósido con acción confirmada en pacientes con enfermedad linfoblástica de células T en recaída y resistente al tratamiento) en el contexto de un régimen del BFM para los pacientes con LLA-T recién diagnosticada.
El uso de la radioterapia craneal profiláctica en el tratamiento de la LLA-T está disminuyendo. Algunos grupos, como el St. Jude Children's Research Hospital (SJCRH) y el Dutch Childhood Oncology Group (DCOG), no utilizan la radioterapia craneal en el tratamiento de primera línea de la LLA. Otros grupos, como el DFCI, el COG y el BFM, ahora limitan la radioterapia a los pacientes con características de riesgo muy alto o enfermedad SNC3.
Opciones de tratamiento en evaluación clínica para la leucemia linfoblástica aguda de células T
La información en inglés sobre los ensayos clínicos patrocinados por el NCI se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Lactantes con leucemia linfoblástica aguda
La leucemia linfoblástica aguda (LLA) es poco frecuente en los lactantes y representa cerca del 2 % al 4 % de los casos de LLA infantil. Debido a sus características biológicas distintivas y el riesgo alto de recidivas leucémicas, los lactantes con LLA se tratan con protocolos diseñados de manera específica para esta población de pacientes. Los aspectos terapéuticos comunes de los regímenes quimioterapéuticos intensivos que se usanpara los lactantes con LLA son la adición de cursos de intensificación durante la posinducción con dosis altas de citarabina y metotrexato.
El desenlace es especialmente precario cuando se establece el diagnóstico durante los primeros meses de vida del lactante. En un estudio, la tasa de SG a 2 años fue del 20 % cuando el diagnóstico se hizo durante el primer mes de vida.[Nivel de evidencia B4] En otro estudio, la tasa de SSC a 5 años para los lactantes que recibieron el diagnóstico durante los primeros 90 días de vida fue del 16 %.[Nivel de evidencia B4]
En los lactantes con reordenamientos del gen KMT2A, las tasas de SSC a los 4 a 5 años son de alrededor del 35 %.[Nivel de evidencia B4] Los factores pronósticos de un desenlace precario en los lactantes con reordenamientos de KMT2A son los siguientes:; [Nivel de evidencia C2]; [Nivel de evidencia B4]
- Edad más joven en el momento del diagnóstico (≤90–180 días).
- Recuento leucocitario demasiado alto en el momento de la presentación inicial (≥200 000–300 000/μl).
- Respuesta temprana precaria, que se ve reflejada en una respuesta precaria a la profase de prednisona o índices altos de enfermedad residual mínima (ERM) al final de las fases de inducción y consolidación del tratamiento.
En un informe, también se encontró que la presencia de compromiso en el SNC de cualquier grado en el momento del diagnóstico (SNC2, SNC3, o punción lumbar traumática con blastocitos) era un factor pronóstico independiente de desenlace precario en lactantes con LLA con reordenamiento de KMT2A.
Además de tener una tasa de recaída significativamente más alta que los niños de más edad con LLA, los lactantes suelen presentar más a menudo una enfermedad de mayor gravedad. En un estudio retrospectivo de gran tamaño, los lactantes con LLA fueron más propensos a presentar insuficiencia multiorgánica que los niños mayores (12 % y 1 %, respectivamente). Los lactantes también tuvieron mayores requerimientos de hemoderivados, diuréticos, oxígeno complementario y ventilación mecánica durante la inducción, en comparación con los niños mayores.
Los lactantes también tienen un mayor riesgo de presentar efectos tóxicos relacionados con el tratamiento, en especial infecciones. Con los abordajes de tratamiento vigentes para esta población, se notifica una mortalidad relacionada con el tratamiento de alrededor del 10 % en los lactantes, una tasa mucho más alta que la de los niños de más edad con LLA. En el ensayo del COG AALL0631 (NCT00557193) , un régimen de inducción intensificado produjo una tasa de mortalidad durante la inducción del 15,4 % (4 de 26 pacientes). Más tarde, el ensayo se modificó con una inducción menos intensiva y mejores directrices para la atención de apoyo, lo que condujo a una tasa de mortalidad durante la inducción significativamente inferior (1,6 %; 2 de 123 pacientes) y una tasa de RC significativamente superior (94 %) que la que se obtuvo con el régimen anterior de inducción más intensiva (68 %).
Opciones de tratamiento para los lactantes con reordenamientos de KMT2A
Los lactantes con reordenamientos del gen KMT2A por lo general se tratan con regímenes quimioterapéuticos intensificados que incluyen fármacos que no se suelen usar en la terapia de primera línea para los niños con LLA de más edad. Sin embargo, a pesar de los abordajes intensificados, las tasas de SSC siguen siendo precarias en estos pacientes.
Evidencia (regímenes quimioterapéuticos intensificados para los lactantes con reordenamientos de KMT2A):
- En el ensayo internacional Interfant 99 se usó un régimen quimioterapéutico intensivo de citarabina con una mayor exposición a dosis bajas y altas de citarabina durante los primeros meses de tratamiento.
- La SSC a 5 años de los lactantes con reordenamientos de KMT2A fue del 37 %.
- El COG evaluó la intensificación del tratamiento con un régimen que incluía múltiples dosificaciones de dosis altas de metotrexato, ciclofosfamida y etopósido.
- La SSC a 5 años de los lactantes con reordenamientos de KMT2A fue del 34 %.
- En el ensayo del COG P9407 (NCT00002756) , se trató a los lactantes con un régimen de quimioterapia intensiva más breve (46 semanas).[Nivel de evidencia B4]
- La SSC a 5 años de los lactantes con reordenamientos de KMT2A fue del 36%.
- En el estudio internacional Interfant 06 se probó si el estilo de quimioterapia de consolidación que se usa para la leucemia mieloide aguda (LMA) era superior al estilo de quimioterapia que se usa para la LLA.[Nivel de evidencia B4]
- La tasa de SSC a 6 años fue del 46,1 %, y la tasa de SG fue del 58,2 %. Estas tasas no presentaron diferencias significativas con las tasas que se observaron en el protocolo anterior Interfant-99.
- En los lactantes con reordenamientos de KMT2A, la SSC a 6 años fue del 36,4 %, sin diferencias significativas entre el abordaje de la LMA y la LLA.
- En un análisis posterior de los subgrupos de pacientes con reordenamiento en KMT2A en los que se evaluó la ERM, se observó que la ERM al final de la consolidación y la ERM al final de la inducción, eran factores pronóstico importantes del desenlace. Los pacientes con ERM positiva al final de la inducción, pero que lograban la ERM negativa al final de la consolidación, tenían desenlaces similares a los de aquellos con ERM al final de la inducción (tasas de SSE a 6 años, 65,7 y 72,0 %, respectivamente). Los pacientes con ERM alta al final de la consolidación presentaron resultados desalentadores (tasa de SSE a 6 años, 13,1 %). Los pacientes con ERM negativa al final de la inducción tuvieron una tasa de SSE a 6 años más alta cuando se trataron con el abordaje para LLA (78,2 %) que la de los pacientes que se trataron con un abordaje para LMA (45 %). Sin embargo, los pacientes con ERM alta al final de la inducción (≥ 5 × 10-4) que se trataron con un abordaje para LMA tuvieron una tasa superior de SSE a 6 años (45,9 %) cuando se la comparó con la de los pacientes que se trataron con un abordaje para LLA (23,2 %).[Nivel de evidencia B1]
- En un ensayo piloto de seguimiento que condujo el grupo de estudio Interfant, 30 lactantes con LLA con reordenamientos en KMT2A se trataron con la pauta quimioterapéutica de base Interfant-06, a lo que se añadió un curso de 28 días de blinatumomab después de la fase de inducción.
- El blinatumomab se toleró bien, sin toxicidad excesiva o inesperada en estos pacientes lactantes.
- Al cabo de una mediana de seguimiento corta (26,3 meses), la tasa de SSE a 2 años fue del 81,6 % (IC 95 %, 60,8–92,0 %), y la tasa de SG a 2 años fue del 93,3% (IC 95 %, 75,9–98,3 %).
- Estos desenlaces fueron superiores a las tasas de SSE y SG a 2 años de los controles históricos que se trataron en el protocolo Interfant-06 (tasas de SSE y SG, 49,4 % y 65,8 %, respectivamente).
- En el ensayo MLL-10 que llevó adelante el Japanese Pediatric Leukemia/Lymphoma Study Group (JPLSG), los lactantes con reordenamientos de KMT2A se trataron con una quimioterapia de base intensificada que incluyó múltiples fases con dosis altas de metotrexato, ciclofosfamida, etopósido y dosis altas de citarabina. Los pacientes que se clasificaron como de riesgo alto según la edad y el estado del SNC (75 % de los pacientes con reordenamiento de KMT2A) se asignaron para recibir un TCMH en la primera RC.
- La tasa de SSC a 5 años fue del 66 % y la tasa de SG a 5 años fue del 82 %.
- En el estudio del COG AALL0631 (NCT00557193), los lactantes con LLA que presentaban reordenamiento de KMT2A se asignaron a recibir un régimen quimioterapéutico intensivo con lestaurtinib, un inhibidor de FLT3 (administrado durante las fases de posinducción), o sin este.
- En todos los participantes, la tasa de SSC a 6 años fue del 34 % y la tasa de SG a 5 años fue del 41 %.
- No hubo diferencias en los desenlaces entre los pacientes del grupo de lestaurtinib versus aquellos que solo recibieron quimioterapia.
Se realizaron estudios exploratorios para evaluar el efecto de las concentraciones de lestaurtinib en sangre suficientes como para lograr la inhibición de FLT3, y para evaluar el efecto de la sensibilidad ex vivo de las células leucémicas al lestaurtinib.
- Solo se demostró la inhibición in vivo de FLT3 en el 38 % de los pacientes tratados con lestaurtinib. Este subgrupo de pacientes tiene un desenlace superior al de otros pacientes en el grupo de lestaurtinib, al igual que aquellos cuyos blastocitos presentaron sensibilidad ex vivo a la inhibición de FLT3.
La función del TCMH alogénico durante la primera remisión en los lactantes con reordenamientos del gen KMT2A continúa siendo polémica.
Evidencia (TCMH alogénico durante la primera remisión en lactantes con reordenamientos de KMT2A):
- En un ensayo clínico que realizó el JPLSG entre 1998 y 2002, se intentó que todos los lactantes con reordenamientos de KMT2A recibieran un TCMH alogénico del mejor donante disponible (emparentado, no emparentado o de cordón umbilical) 3 a 5 meses después del diagnóstico.
- La tasa de SSC a 3 años para todos los lactantes inscritos fue del 44 %. Este desenlace se debió, en parte, a la frecuencia alta de recaídas tempranas, incluso con la quimioterapia intensiva. En ese estudio, de los 41 lactantes con reordenamientos de KMT2A que alcanzaron la RC, 11 (27 %) presentaron recaída antes de someterse al trasplante.
- En un ensayo de seguimiento que realizó el JPLSG entre 2011 y 2015, solo se asignó a TCMH en la primera RC a los pacientes con reordenamientos de KMT2A y características de riesgo alto (edad <6 meses o estado SNC3 en el momento del diagnóstico).
- De los 56 pacientes que se clasificaron como de riesgo alto (75 % de todos los pacientes con reordenamiento de KMT2A incluidos en el ensayo), 49 alcanzaron la RC y 38 se sometieron a TCMH.
- En un análisis por intención de tratar de todos los pacientes de riesgo alto incluidos en el ensayo, la tasa de SSC a 5 años fue del 56,6 %.
- En el subgrupo de pacientes que también cumplía con los criterios de riesgo alto de Interfant-06 (edad <6 meses y recuento de GB >300 000/µl o una respuesta insuficiente a la prednisolona), la tasa de SSC a 5 años fue del 45,2 %.
- En un informe del COG que incluyó a 189 lactantes tratados entre 1996 y 2000 con protocolos del CCG o del POG para lactantes con LLA, no se encontraron diferencias en la SSC entre los pacientes que se sometieron a un TCMH en la primera RC y los que recibieron quimioterapia sola.
- El grupo de ensayos clínicos Interfant, después de hacer un ajuste por tiempo de espera para el trasplante, tampoco observó diferencia en la SSE en lactantes de riesgo alto (definidos por respuesta a la prednisona) con reordenamientos de KMT2A tratados en el ensayo Interfant 99 con un TCMH alogénico en la primera RC o quimioterapia sola.
- En un análisis de subconjuntos del mismo ensayo, el TCMH alogénico durante la primera remisión se relacionó con una SSE significativamente superior para los lactantes con reordenamientos de KMT2A que tenían menos de 6 meses en el momento del diagnóstico y que presentaron una respuesta precaria a la prednisona en el día 8, o un recuento leucocitario de por lo menos 300 000/µl. En este subconjunto, el TCMH en la primera remisión se relacionó con una reducción del 64 % en el riesgo de fracaso por recaída o muerte en comparación con el grupo de quimioterapia sola.
- En el estudio Interfant 06, los lactantes clasificados en el grupo de riesgo alto (que cumplían con todas las características siguientes: reordenamientos de KMT2A, edad <6 meses, y GB ≥300 000/μl) se consideraron aptos para recibir un TCMH alogénico en el momento de la primera RC.[Nivel de evidencia B4]
- Casi la mitad de los pacientes de riesgo alto no se sometieron a un trasplante en el momento de la primera RC debido, sobre todo, a una recaída temprana.
- En todo el grupo de riesgo alto, la tasa de SSC a 6 años fue del 21 %.
- Para la población de pacientes muy seleccionados que se sometieron a un TCMH, la tasa de SSE a 4 años fue del 44 %.
En los lactantes con LLA sometidos a trasplante después de la primera RC, los desenlaces de los que reciben regímenes con irradiación corporal total (ICT) son semejantes a los de aquellos que no reciben ICT.
Opciones de tratamiento para los lactantes sin reordenamientos de KMT2A
El tratamiento óptimo para los lactantes sin reordenamientos de KMT2A tampoco está claro, en parte debido a la escasez de datos sobre el uso de los regímenes de LLA estándar que se emplean en niños de más edad.
- En el ensayo Interfant 99, los pacientes sin reordenamientos de KMT2A lograron un resultado relativamente favorable y comparable al del régimen de tratamiento intensivo a base de citarabina (tasa de SSC a 4 años, 74 %).
- En el ensayo del COG P9407 (NCT00002756) de quimioterapia intensificada, se notificó una tasa de SSC a 5 años del 70 % en lactantes sin el reordenamiento de KMT2A.[Nivel de evidencia B4]
- Se obtuvo un resultado favorable para este subgrupo de pacientes en un estudio japonés en el que se usó un tratamiento comparable al que se utiliza para los niños con LLA de más edad; sin embargo, el estudio estuvo limitado por su número reducido (n = 22) y una distribución por sexo muy poco habitual (91 % eran hombres).
- En el estudio Interfant 06, la tasa de SSC a 6 años para los lactantes sin reordenamientos de KMT2A fue del 73,9 %, y la tasa de SG fue del 87,2 %.; [Nivel de evidencia B4]
- En el ensayo del COG AALL0631 (NCT00557193) , los lactantes sin reordenamientos de KMT2A recibieron la misma quimioterapia de base intensificada que aquellos con reordenamientos de KMT2A.
- La tasa de SSC a 5 años del 87,3 % (± 4.7%), y la tasa de SG a 5 años fue del 93,6 % (± 3,5 %) en 64 lactantes sin reordenamientos de KMT2A.
Opciones de tratamiento en evaluación clínica para los lactantes con leucemia linfoblástica aguda
La información en inglés sobre los ensayos clínicos patrocinados por el NCI se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
Adolescentes y adultos jóvenes con leucemia linfoblástica aguda
Desde hace décadas los adolescentes y adultos jóvenes con leucemia linfoblástica aguda (LLA) se han considerado de riesgo alto. Los desenlaces en este grupo etario son inferiores en casi todos los estudios de tratamiento en comparación con los de los niños menores de 10 años. Esta diferencia se debe a que en el momento del diagnóstico estos pacientes presentan más a menudo factores de pronóstico adverso, como los siguientes:
- Inmunofenotipo de células T.
- BCR::ABL1 y enfermedad similar a BCR::ABL1.
- Incidencia más baja de anomalías citogenéticas favorables.
Además de los factores pronósticos adversos más frecuentes, los pacientes de este grupo etario tienen tasas más altas de mortalidad relacionada con el tratamiento y de incumplimiento terapéutico.
Opciones de tratamiento para los adolescentes y adultos jóvenes con leucemia linfoblástica aguda
En algunos estudios de los Estados Unidos y Francia se identificaron por primera vez las diferencias en los desenlaces de acuerdo con los regímenes de tratamiento. En otros estudios se confirmó que los pacientes adolescentes mayores y adultos jóvenes presentan una evolución más favorable cuando reciben regímenes para niños en lugar de regímenes para adultos.; [Nivel de evidencia B4] Estos resultados del estudio se resumen en el Cuadro 12.
Debido al resultado relativamente favorable que se obtiene en estos pacientes cuando reciben los regímenes quimioterapéuticos usados para la LLA infantil de riesgo alto, no hay lugar para el uso rutinario de un TCMH alogénico en el momento de la primera remisión de los adolescentes y adultos jóvenes con LLA.
Evidencia (uso de un régimen de tratamiento pediátrico en adolescentes y adultos jóvenes con LLA):
- En el ensayo CALGB-10403 (NCT00558519) se estudió la viabilidad y la eficacia de usar un régimen de tratamiento pediátrico (administrado por oncólogos médicos) para pacientes adolescentes y adultos jóvenes con LLA recién diagnosticada. De los 318 pacientes inscritos, 295 fueron aptos y evaluables para la respuesta. La mediana de edad fue de 24 años (intervalo de edad de 17–39 años).
- El uso de un régimen pediátrico (del estudio COG AALL0232, de dosis escalonadas de metotrexato sin leucovorina seguido de asparaginasa) se consideró inocuo y la mortalidad general relacionada con el tratamiento fue del 3 %.
- La mediana de SSC fue de 78,1 meses, más del doble del control histórico de 30 meses. La tasa de SSC a 3 años fue del 59 % y no se alcanzó la mediana de SG. La tasa estimada de SG a 3 años fue del 73 %.
- Los factores de riesgo anteriores al tratamiento relacionados con un desenlace más precario fueron la obesidad y la presencia de la firma de expresión similar a BCR::ABL1. De los pacientes evaluables, el 31 % presentó la firma de expresión similar a BCR::ABL1. Estos pacientes presentaron un desenlace mucho más precario, con una tasa de SSC a 3 años del 42 %, en comparación con una tasa de SSC del 69 % en los pacientes con LLA similar a BCR::ABL1 (cociente de riesgos instantáneos, 2,06; orden logarítmico, P = 0,008).
- La ERM al final de la inducción fue un factor pronóstico importante del desenlace. En el subgrupo de pacientes evaluables para la ERM, en el 44 % no se observó ERM detectable al final de la inducción mediante una prueba de PCR de Ig y receptor de célula T (sensibilidad de detección de 1 en 104 a 105). Estos pacientes tuvieron una tasa de SSE a 3 años del 85 %, en comparación con una tasa de SSE a 3 años del 54 % en aquellos con MRD detectable al final de la inducción (P = 0,001).
- En un estudio de pacientes de 16 a 39 años de edad, se compararon los desenlaces de aquellos que recibieron quimioterapia estándar de tipo pediátrico posremisión según el protocolo CALGB-10403 con los resultados de los pacientes que recibieron TCMH alogénico mielosupresor. Los pacientes que recibieron quimioterapia tuvieron tasas superiores de SG, SSE y mortalidad no relacionada con las recaídas.[Nivel de evidencia B4]
- Los investigadores informaron sobre 197 pacientes de 16 a 21 años tratados en el estudio del CCG (régimen para la LLA infantil) que se compararon con 124 adolescentes y adultos jóvenes tratados en el estudio Cancer and Leukemia Group B (CALGB) (régimen para la LLA en adultos).
- En los pacientes tratados con un régimen para la LLA infantil, la tasa de SSC a 7 años fue del 63 %.
- En los pacientes tratados con un régimen para la LLA en adultos, la tasa de SSC a 7 años fue del 34 %.
- En un estudio de cohortes poblacional canadiense, se determinó el efecto de adaptar los protocolos pediátricos para usarlos en pacientes adolescentes y adultos jóvenes con LLA durante un periodo de 20 años.
- La tasa de SSC a 5 años para los pacientes adolescentes y adultos jóvenes tratados en centros pediátricos fue del 72 %, en comparación con una tasa de SSC del 56 % en los pacientes adolescentes y adultos jóvenes tratados en centros para adultos (P = 0,03).
- En el periodo más reciente (2006–2011), el desenlace de los pacientes adolescentes y adultos jóvenes tratados con protocolos pediátricos en centros para adultos fue superior al desenlace de los pacientes tratados con protocolos para adultos (tasa de SSC, 72 vs. 60 %), pero inferior al de los pacientes adolescentes y adultos jóvenes tratados en centros pediátricos (tasa de SSC, 81 %; P = 0,02).
- Los autores concluyen que, además del protocolo de tratamiento, quizás haya otras diferencias entre los centros pediátricos y de adultos que expliquen la disparidad en los desenlaces.
Se desconoce la razón por la que los adolescentes y adultos jóvenes logran resultados superiores con los regímenes pediátricos, aunque las posibles explicaciones comprenden las siguientes:
- Entorno del tratamiento (es decir, la experiencia del centro en el tratamiento de la LLA).
- Cumplimiento con el protocolo de tratamiento.
- Componentes del protocolo de tratamiento.
| Sitio y grupo de estudio | Pacientes adolescentes y adultos jóvenes (No.) | Mediana de edad (años) | Supervivencia (%) |
|---|---|---|---|
| LLA = Leucemia linfoblástica aguda; SSC = supervivencia sin complicaciones; SG = supervivencia general. | |||
| AIEOP = Associazione Italiana di Ematologia e Oncologia Pediatrica; CALGB = Cancer and Leukemia Group B; CCG = Children's Cancer Group; DCOG = Dutch Childhood Oncology Group; FRALLE = French Acute Lymphoblastic Leukaemia Study Group; GIMEMA = Gruppo Italiano Malattie EMatologiche dell'Adulto; HOVON = Dutch-Belgian Hemato-Oncology Cooperative Group; LALA = France-Belgium Group for Lymphoblastic Acute Leukemia in Adults; MRC = Medical Research Council (United Kingdom); NOPHO = Nordic Society for Pediatric Hematology and Oncology; UKALL = United Kingdom Acute Lymphoblastic Leukaemia. | |||
| Estados Unidos | |||
| CCG (pediátrico) | 197 | 16 | 67, SG 7 años |
| CALGB (adultos) | 124 | 19 | 46 |
| Francia | |||
| FRALLE 93 (pediátrico) | 77 | 16 | 67 SSC |
| LALA 94 | 100 | 18 | 41 |
| Italia | |||
| AIEOP (pediátrico) | 150 | 15 | 80, SG 2 años |
| GIMEMA (adultos) | 95 | 16 | 71 |
| Países Bajos | |||
| DCOG (pediátrico) | 47 | 12 | 71 SSC |
| HOVON | 44 | 20 | 38 |
| Suecia | |||
| NOPHO 92 (pediátrico) | 36 | 16 | 74, SG 5 años |
| LLA en adultos | 99 | 18 | 39 |
| Reino Unido | |||
| MRC LLA (pediátrico) | 61 | 15–17 | 71, SG 5 años |
| UKALL XII (adultos) | 67 | 15–17 | 56 |
| UKALL 2003 | 229 | 16–24 | 72 SSC |
Osteonecrosis
Los adolescentes con LLA tienen un riesgo más alto que los niños pequeños de presentar complicaciones relacionadas con el tratamiento, como osteonecrosis, trombosis venosa profunda y pancreatitis. Antes de la intensificación de la posinducción para el tratamiento de la LLA, la osteonecrosis era poco frecuente. La mejora de los resultados en niños y adolescentes de 10 años o más estuvo acompañada por un aumento de la incidencia de osteonecrosis.
En el 95 % de los pacientes con osteonecrosis, se ven afectadas las articulaciones que soportan peso y, en más del 40 % de los casos, se necesitaron intervenciones quirúrgicas con el fin de controlar los síntomas y el deterioro de la movilidad. El diagnóstico en la mayoría de los casos se establece durante los primeros 2 años de tratamiento y, con frecuencia, los síntomas se identifican durante el mantenimiento.
Evidencia (osteonecrosis):
- En el estudio CCG-1961 de LLA de riesgo alto el uso de una dosificación de dexametasona en semanas alternas se comparó con la administración de dexametasona continua estándar durante la intensificación diferida a fin de determinar si se podía reducir el riesgo de osteonecrosis.
- La mediana de edad de inicio de los síntomas fue de 16 años.
- La incidencia acumulada fue más alta en los adolescentes y los adultos jóvenes de 16 a 21 años (20 % a los 5 años) que en aquellos de 10 a 15 años (9,9 %) o en pacientes de 1 a 9 años (1 %).
- Se necesitaron intervenciones quirúrgicas para controlar los síntomas y las alteraciones de movilidad en más del 40 % de los casos.
- En el estudio CCG-1961, el uso de una dosificación de dexametasona en semanas alternas en comparación con dexametasona continua estándar durante la intensificación diferida redujo el riesgo de osteonecrosis. El mayor efecto se observó en las jóvenes de 16 a 21 años que presentaron la incidencia más alta de osteonecrosis con la terapia estándar de dexametasona continua; la osteonecrosis se redujo con dexametasona en semanas alternas durante la posinducción (del 57,6 al 5,6 %).
- En el estudio del COG AALL0232 (NCT00075725) de leucemia linfoblástica aguda de riesgo alto, los pacientes se asignaron al azar durante la inducción a recibir 14 días de dexametasona o 28 días de prednisona.
- La incidencia de osteonecrosis en pacientes mayores de 10 años que recibieron dexametasona fue del 24,3 %, en comparación con una incidencia del 15,9 % en aquellos que recibieron prednisona (P = 0,001)
- En los pacientes de 10 años o más, no hubo diferencias en el desenlace entre los grupos por corticoesteroide (tasa de SSC a 5 años, 73,1 % en el grupo de dexametasona y 73,9 % en el grupo de prednisona; P = 0,78).
Opciones de tratamiento en evaluación clínica para los pacientes adolescentes y adultos jóvenes con leucemia linfoblástica aguda
La información en inglés sobre los ensayos clínicos patrocinados por el NCI se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presentan ejemplos de ensayos clínicos nacionales o institucionales en curso:
- A041501 (NCT03150693) (Inotuzumab Ozogamicin and Frontline Chemotherapy in Treating Young Adults With Newly Diagnosed B-cell ALL): este es un ensayo de la National Clinical Trials Network cuyo objetivo es ampliar la experiencia del uso de una quimioterapia de base inspirada en la práctica pediátrica para los adultos jóvenes con LLA. Los criterios de elegibilidad incluyen a pacientes de 18 a 39 años con diagnóstico reciente de LLA positiva para CD22. Los pacientes que estén en remisión después de la inducción se asignarán al azar a recibir el tratamiento de base pediátrico con 2 cursos de inotuzumab ozogamicina (un conjugado de anticuerpo monoclonal anti-CD22 y una toxina) o sin estos antes de comenzar la terapia de consolidación.
- COG-AALL1721 (NCT03876769) (Study of Efficacy and Safety of Tisagenlecleucel in High-Risk B-ALL End-of-Consolidation MRD-Positive Patients): el objetivo del estudio es evaluar la eficacia de la terapia de células T con un receptor del antígeno quimérico (CAR) de CD19 (tisagenlecleucel) en pacientes con ERM al final de la consolidación según la medición de la SSC a 5 años. Otros objetivos incluyen la evaluación de la proporción de personas que no tienen enfermedad y no reciben trasplante alogénico al cabo de 1 año, la SG y la proporción de personas que logran una RC negativa para la ERM o una RCI a los 3 meses del tisagenlecleucel.
- COG-AALL1732 (NCT03959085) (A Phase III Randomized Trial of Inotuzumab Ozogamicin for Newly Diagnosed High-Risk B-ALL; Risk-Adapted Postinduction Therapy for High-Risk B-ALL, Mixed Phenotype Acute Leukemia [MPAL], and Disseminated B-Lymphoblastic Lymphoma): este protocolo está abierto para pacientes menores de 25 años de edad en el momento del diagnóstico de cualquiera de las siguientes afecciones: LLA-B de riesgo alto del NCI en paciente sin síndrome de Down, LAFM y linfoma linfoblástico de células B diseminado. En el protocolo se evalúa si la adición de 2 bloques de inotuzumab ozogamicina a un tratamiento de base del BFM modificado para pacientes con LLA-B mejorará la SSE. Para los pacientes con LAFM y linfoma linfoblástico de células B diseminado, el estudio procura determinar la SSC relacionada con un tratamiento de base del BFM modificado para la LLA-B de riesgo alto estándar.
Niños con síndrome de Down
Alrededor de un 2 % a un 3 % de los casos de LLA se presentan en niños con síndrome de Down. La LLA de pacientes pediátricos con síndrome de Down se caracteriza por una incidencia baja de características biológicas favorables (por ejemplo, ETV6::RUNX1 e hiperdiploidía alta con trisomías favorables) y desfavorables (por ejemplo, BCR::ABL1, reordenamientos de KMT2A, hipodiploidía baja, t(9;22)(q34;q11.2) o t(4;11)(q21;q23)), así como una ausencia casi total de fenotipo de células T.
Determinadas anomalías genómicas, como las deleciones de IKZF1, las anomalías de CRLF2 y las variantes de JAK se observan con mayor frecuencia en la LLA de los niños con síndrome de Down que en aquellos sin este síndrome. Los estudios de niños con síndrome de Down y LLA indican que la presencia de deleciones de IKZF1 (pero no anomalías de CRLF2 o variantes de JAK) acarrea un pronóstico más precario.
Los pacientes con síndrome de Down tienen un mayor riesgo de presentar efectos tóxicos relacionados con el tratamiento, como infecciones, mucositis y convulsiones. En algunos estudios, se notificaron desenlaces más precarios en niños con síndrome de Down y LLA, sin embargo, en otros estudios, los pacientes con síndrome de Down evolucionaron tan bien como los que no tenían este síndrome. Cuando se observan desenlaces inferiores en los pacientes con síndrome de Down, se relacionan con un mayor riesgo de recaída y una mayor frecuencia de mortalidad relacionada con el tratamiento.
Debido al aumento comprobado de la toxicidad que presentan los pacientes con síndrome de Down, algunos protocolos de LLA (como los del COG) han desintensificado el tratamiento según el riesgo en los pacientes con síndrome de Down y LLA para minimizar la exposición a los componentes de la terapia que producen morbilidad. Si bien esta estrategia de reducción del tratamiento disminuye la frecuencia y los efectos tóxicos, su efecto en los resultados antileucémicos todavía se desconocen.
Tratamiento de los niños con síndrome de Down
Evidencia (efectos tóxicos y desenlace de los pacientes con síndrome de Down y LLA):
- En un estudio retrospectivo grande en el que se incluyeron 653 pacientes con síndrome de Down y LLA tratados entre 1995 y 2004 se obtuvieron los siguientes resultados:
- Los pacientes con síndrome de Down tuvieron tasas de RC más bajas (97 vs. 99 %, P< 0,001), tasas más altas de incidencia acumulada de recaída (26 vs. 15 %, P< 0,001) y tasas más altas de mortalidad relacionada con el tratamiento (7,7 vs. 2,3 %, P< 0,001), en comparación con los pacientes sin este síndrome.
- En los pacientes con síndrome de Down y LLA, la tasa de mortalidad relacionada con el tratamiento fue más alta en todas las fases de tratamiento, incluso en el mantenimiento. Las tasas de mortalidad relacionada con el tratamiento durante la inducción fueron del 2,8 % y del 4,9 % durante el resto de las fases de tratamiento, una vez que se alcanzó la RC. La causa más frecuente de mortalidad relacionada con el tratamiento fue la infección.
- En los pacientes con síndrome de Down, la edad inferior a 6 años, el recuento de GB inferior a 10 000/µl y la fusión ETV6::RUNX1 (que se observó en un 8 % de los pacientes) fueron factores de predicción independientes de una SSC favorable.
- En un informe de 2811 niños con LLA que participaron en el estudio de clasificación COG P9900, 80 pacientes (3 %) tenían síndrome de Down. La edad, el sexo, el recuento de GB y el grupo de riesgo fueron similares entre los pacientes con síndrome de Down y sin este síndrome, pero un porcentaje menor de pacientes con síndrome de Down presentaron ETV6::RUNX1 (2,5 vs. 24 %, P< 0,001) o trisomías 4 y 10 (7,7 vs. 24 %, P< 0,001).
- Las tasas de SSC y SG a 5 años fueron inferiores en los niños con síndrome de Down: 69,9 % versus 78,1 % (P = 0,078) y 85,8 % versus 90,0 % (P = 0,033), respectivamente.
- Sin embargo, cuando los niños con reordenamientos de KMT2A, BCR::ABL1, ETV6::RUNX1 y trisomías 4 y 10 se excluyeron del análisis, las tasas de SSC y SG fueron similares entre los niños con síndrome de Down y los que no tenían el síndrome (tasas de SSC, 68,0 % [± 9,3 %] vs. 70,5 % [± 1,9 %], P = 0,817; tasas de SG, 86,7% [± 6,7 %] vs. 85,4 % [± 1,5 %]; P = 0,852).
- En el estudio CCG-1991 (NCT00005945) de niños con LLA-B de riesgo estándar (2000–2005), los pacientes (incluso aquellos con síndrome de Down) se asignaron al azar para recibir una fase de mantenimiento provisional que consistió en dosis escalonadas de metotrexato intravenoso con rescate de leucovorina o sin este, o dosis bajas estándar de metotrexato oral con vincristina, dexametasona y mercaptopurina.
- Las tasas de SSC a 10 años fueron del 94,4 % (± 5,4 %) en los pacientes con síndrome de Down que se asignaron al azar para recibir metotrexato IV en dosis escalonadas (N = 31), versus el 81,5 % (± 6,6 %) en los pacientes que recibieron dosis bajas de metotrexato oral (N = 44).
- La dosis media tolerada de metotrexato en el grupo de dosis escalonadas IV fue más baja en los pacientes con síndrome de Down que en los pacientes sin el síndrome.
- La incidencia de mucositis aumentó en los pacientes con síndrome de Down que recibieron metotrexato en dosis escalonadas, en comparación con los pacientes sin síndrome de Down, pero no hubo ningún incremento de toxicidad hepática, infecciones o muertes relacionadas con el tratamiento.
- En los protocolos del DFCI ALL Consortium, los niños con síndrome de Down reciben el mismo tratamiento estratificado que los demás pacientes, sin reducciones ni modificaciones de las dosis. En un análisis de 2 ensayos consecutivos realizados entre 2000 y 2011 se observó lo siguiente:
- Las tasas de SSC y SG a 5 años de los pacientes con síndrome de Down (N = 38) fueron similares a las de aquellos sin síndrome de Down (N = 1248) (tasas de SSC, 91 vs. 84 %; tasas de SG, 97 vs. 91 %).
- Todos los pacientes con síndrome de Down lograron la remisión completa y no hubo muertes relacionadas con el tratamiento.
- Los pacientes con síndrome de Down tuvieron tasas significativamente más altas de mucositis (52 vs. 12 %, P< 0,001), trombosis que no afectaron el SNC (18 vs. 8 %; P = 0,036) y convulsiones (16 vs. 5 %, P = 0,010). Los pacientes con síndrome de Down también tuvieron una incidencia más alta de infecciones durante todas las fases de tratamiento.
Opciones de tratamiento en evaluación clínica para los niños con síndrome de Down y leucemia linfoblástica aguda
La información en inglés sobre los ensayos clínicos patrocinados por el NCI se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de un ensayo clínico nacional o institucional en curso:
- COG-AALL1731 (NCT03914625) (A Phase III Trial Investigating Blinatumomab in Combination with Chemotherapy in Patients with Newly Diagnosed Standard Risk or Down Syndrome B-ALL and the Treatment of Patients with Localized B-Lymphoblastic Lymphoma): en este estudio se está evaluando si la adición de blinatumomab a la quimioterapia estándar mejora la SSE. Todos los pacientes con síndrome de Down (incluso los adolescentes y adultos jóvenes <31 años de edad) son aptos para la inscripción. Los pacientes con riesgo estándar del NCI y síndrome de Down que cumplen con la definición de riesgo estándar promedio recibirán el mismo tratamiento que los pacientes sin síndrome de Down que presentan riesgo estándar promedio. Esos pacientes son aptos para participar en la aleatorización del blinatumomab. El resto de los pacientes con síndrome de Down, incluso los de riesgo alto del NCI, características biológicas desfavorables y ERM alta el día 29 se clasificarán en el grupo de síndrome de Down de riesgo alto y se asignarán al azar a recibir 2 ciclos de blinatumomab, además del régimen de quimioterapia reducido en el que se omiten los elementos intensivos del tratamiento de base del BFM intensificado. Los elementos que se omiten son las antraciclinas durante la inducción y la quimioterapia a base de ciclofosfamida y citarabina durante la segunda parte de la intensificación diferida.
Leucemia linfoblástica aguda positiva para BCR::ABL1 (positiva para el cromosoma Filadelfia)
La leucemia linfoblástica aguda (LLA) positiva para BCR::ABL1 (positiva para el cromosoma Filadelfia [Ph+] se observa en alrededor del 3 % de los casos de LLA infantil, aumenta en la adolescencia y representa un 15 % a un 25 % de los casos de LLA en adultos. En el pasado, se consideraba que este subtipo de LLA era muy difícil de tratar y los pacientes presentaban un desenlace precario. En el año 2000, un grupo internacional de leucemia infantil internacional notificó una tasa de SSC a 7 años del 25 % con una SG del 36 %. En 2010, el mismo grupo notificó una tasa de SSC a 7 años del 31 % y una tasa de SG del 44 % en los pacientes con LLA BCR::ABL1 tratados sin inhibidores de tirosina–cinasas (ITC). El tratamiento de este subgrupo ha evolucionado desde un énfasis inicial en quimioterapia intensiva hasta el trasplante alogénico de células hematopoyéticas en el momento de una remisión completa (RC) como estándar de atención y, en la actualidad, una combinación de quimioterapia con un ITC; solo un número bajo de pacientes se ha sometido al trasplante alogénico en el momento de la primera RC.
En los pacientes con LLA y fusiones génicas BCR::ABL1, la detección de la ERM a partir de citometría de flujo o detección de los reordenamientos de inmunoglobulinas o del receptor de células T (IG/TCR) mediante reacción en cadena de la polimerasa (PCR) o secuenciación de última generación (NGS) brinda una estimación del pronóstico más confiable que los métodos basados en la cuantificación de los trascriptos o el DNA con la fusión BCR::ABL1 o del DNA. En algunos casos, los transcriptos o el DNA con la fusión BCR::ABL1 persisten en ausencia de ERM detectable mediante citometría de flujo o pruebas de reordenamientos de IG/TCR. Este comportamiento es característico de la LLA con fusión BCR::ABL1 y compromiso de múltiples linajes. El subtipo que afecta varios linajes se diferencia del subtipo más común, la LLA con fusión BCR::ABL1 y compromiso linfoide solo, en donde la fusión BCR::ABL1 se puede encontrar en células B, T o mieloides no afectadas por la LLA, en oposición a los linfoblastos solamente. La persistencia de DNA o RNA con la fusión BCR::ABL1 en la LLA con fusión BCR::ABL1 y compromiso de varios linajes posiblemente indique la presencia de un clon preleucémico residual y no de células leucémicas. Por lo tanto, el término ERM es inapropiado. A partir de un número bajo de pacientes estudiados hasta la fecha, el pronóstico es semejante en adultos y niños de LLA con fusión BCR::ABL1 y compromiso linfoide solo o compromiso de múltiples linajes. Además, la persistencia de la positividad en PCR para la fusión BCR::ABL1 (en ausencia de ERM detectable por otros métodos) no tiene repercusión pronóstica. Por lo tanto, en la LLA con fusión BCR::ABL1, la citometría de flujo, las pruebas de PCR para IG/TCR y las pruebas de NGS son métodos más confiables que la PCR para la fusión BCR::ABL1 en cuanto a la evaluación de la ERM como medida de estratificación del riesgo y para la toma de decisiones de tratamiento. Para obtener más información, consultar la sección Características citogenéticas y genómicas de la leucemia linfoblástica aguda infantil.
Opciones de tratamiento para los pacientes con leucemia linfoblástica aguda positiva para BCR::ABL1
El tratamiento estándar para los pacientes con LLA BCR::ABL1 incluye el uso de un ITC (por ejemplo, imatinib o dasatinib) en combinación con quimioterapia citotóxica, con un TCMH alogénico en la primera RC o sin este.
El mesilato de imatinib es un inhibidor selectivo de la proteína cinasa BCR::ABL1. En estudios de fase I y II de imatinib en monoterapia para niños y adultos con LLA BCR::ABL1 recidivante o resistente al tratamiento se demostraron tasas de respuesta relativamente altas, aunque estas respuestas en general fueron de corta duración.
En ensayos clínicos de adultos y niños con LLA BCR::ABL1 se demostró la viabilidad de la administración de mesilato de imatinib en combinación con quimioterapia multifarmacológica. Se observaron mejores desenlaces en los pacientes con LLA BCR::ABL1 después del TCMH cuando se administró imatinib antes o después del trasplante. En ensayos clínicos también se demostró que muchos pacientes pediátricos con LLA BCR::ABL1 obtendrán una SSC similar si se administra quimioterapia y un ITC en lugar de un trasplante.
El dasatinib, un ITC de segunda generación, también se ha estudiado para el tratamiento de la LLA BCR::ABL1. El dasatinib demostró una actividad importante en el SNC en un modelo murino y en una serie de pacientes con leucemia en el SNC. Los resultados de un ensayo de fase I de dasatinib en pacientes pediátricos indicaron que una dosificación una vez al día se relacionó con un perfil de toxicidad aceptable, con pocos efectos adversos no hematológicos de grado 3 o 4.
Evidencia (inhibidor de tirosina–cinasas):
- En el estudio COG-AALL0031 se evaluó si se podía incorporar el mesilato de imatinib a un régimen quimioterapéutico intensivo para los niños con LLA BCR::ABL1. Los pacientes recibieron mesilato de imatinib y quimioterapia durante la terapia posinducción. Algunos niños recibieron un TCMH alogénico después de 2 ciclos de quimioterapia de consolidación con mesilato de imatinib, mientras que otros recibieron mesilato de imatinib en combinación con quimioterapia durante todas las fases del tratamiento.
- La SSE a 5 años de los 25 pacientes que recibieron quimioterapia intensiva y dosis continuas de mesilato de imatinib fue del 70 % (± 12 %). Estos pacientes presentaron una evolución más favorable que los controles históricos tratados con quimioterapia sola (sin mesilato de imatinib), y un desenlace por lo menos tan bueno como el del resto de los pacientes del ensayo que se sometieron a trasplante alogénico. La tasa de SSE a 5 años fue del 66 % (n = 21) en los pacientes que recibieron un trasplante de un donante fraterno y del 59 % para los que recibieron un trasplante de un donante no emparentado (n = 13).
- Los pacientes con anomalías citogenéticas adicionales tuvieron desenlaces más precarios (P = 0,05).
- En el estudio COG-AALL0622 (NCT00720109) se evaluó el uso de dasatinib (en lugar de imatinib) en combinación con una quimioterapia de base similar a la empleada en el ensayo COG-AALL0031.[Nivel de evidencia B4] En este ensayo, el tratamiento con dasatinib comenzó el día 15 de la inducción, mientras que en el ensayo AALL0031, el tratamiento con imatinib comenzó en la posinducción.
- La adición de un ITC durante la inducción en el ensayo AALL0631 produjo una respuesta temprana superior, en comparación con el ensayo AALL0031 (sin ITC durante la inducción). Al final de la inducción (día 29) la tasa de RC fue del 98 % (n = 59) en el ensayo AALL0622, en comparación con el 89 % (n = 91) en el ensayo AALL0031 (P = 0,01), y las tasas de ERM baja al final de la inducción (<0,01 %) fueron superiores en el ensayo AALL0622 en comparación con el ensayo AALL0031 (59 vs. 25 %; P< 0,001).
- Los desenlaces en ambos ensayos fueron similares: las tasas de SG a 5 años fueron del 81 % y del 86 %, y las tasas de SSE a 5 años fueron del 68 % y del 60 % en el AALL0031 y el AALL0622, respectivamente.
- No se observó toxicidad excesiva con dasatinib.
- En un análisis de subconjuntos en el que se incluyeron los pacientes que tenían muestras diagnósticas conservadas, se identificó la deleción de IKZF1 en el 57 % de los pacientes y se relacionó con tasas inferiores de SSE y SG.
- En el ensayo EsPhALL2004 se probó si el imatinib (administrado de forma discontinua) en el contexto de una quimioterapia intensiva mejoraba el pronóstico de los niños con LLA BCR::ABL1 que, en su mayoría (80 %) recibieron un TCMH alogénico en la primera RC. Los pacientes se clasificaron como de riesgo bajo o alto a partir de las mediciones de la respuesta temprana y el estado de la remisión al final de la inducción. Los pacientes con pronóstico favorable (n = 90) se asignaron al azar a recibir imatinib o a no recibirlo; los pacientes con pronóstico desfavorable (n = 70) se asignaron directamente a recibir imatinib. La interpretación de este estudio es limitada debido a la tasa alta de incumplimiento del tratamiento asignado durante la aleatorización de los pacientes con pronóstico favorable, además del cierre prematuro del ensayo antes de que se alcanzara la meta de inclusión debido a la publicación de los resultados del ensayo COG AALL1131, en el que el imatinib se administró de forma continua con quimioterapia.
- La SSE general de los pacientes tratados en este ensayo es superior a la de los controles históricos y, cuando se hace el análisis según el tratamiento (y no por intención de tratar), los pacientes con pronóstico favorable que recibieron imatinib tuvieron una SSE superior (tasa de SSE a 4 años del 75 % en los pacientes que recibieron imatinib y del 56 % en los pacientes que no recibieron imatinib).
- El ensayo posterior EsPhALL2010 (NCT00287105) fue el resultado de una enmienda al ensayo de 2004, que entre otras cosas, incluyó un inicio más temprano de la terapia con imatinib en el día 15 de la inducción y la administración continua de dosis de imatinib hasta el final de la terapia o 1 año después del trasplante. En una modificación posterior del ensayo también se cambió la indicación del TCMH en el momento de la primera RC solo para los pacientes con pronóstico desfavorable.
- La introducción temprana del imatinib (durante la inducción) produjo un aumento de la tasa de RC hasta el 97 % al final de la inducción (desde el 78 % en el ensayo anterior) y se asignaron menos pacientes al TCMH (38 % en el ensayo modificado vs. 81 % en el ensayo inicial).
- Las tasas de SSE y SG fueron similares en el ensayo inicial y el modificado, aunque un número significativamente más bajo de pacientes recibió el TCMH durante la primera RC en el ensayo modificado.
- La quimioterapia de base de EsPhALL combinada con una dosificación continua de imatinib se relacionó con una tasa alta de efectos tóxicos (sobre todo de infecciones) y de mortalidad relacionada con el tratamiento.
- El estudio CA180-372/COG AALL1122 (NCT01460160) fue un ensayo de fase II y un solo grupo llevado a cabo de manera conjunta por el COG y el intergrupo europeo en donde se evaluó la combinación de dasatinib y quimioterapia de base EsPhALL para los pacientes de LLA con BCR::ABL1 recién diagnosticados. El dasatinib se inició el día 15 de la terapia de inducción y se administró radioterapia craneal solo a los pacientes SNC3 o asignados a TCMH. A 19 pacientes entre 109 (18 %) se les clasificó en el grupo de riesgo alto de acuerdo con una ERM alta en el momento de la segunda medición (final de la inducción 1B) o por ERM detectable (cualquier nivel) después de 3 bloques de consolidación con quimioterapia. Al resto de los pacientes se les asignó el riesgo estándar (82 %). Del grupo de riesgo alto, 15 pacientes (14 %) recibieron TCMH en el momento de la primera RC. El resto de los pacientes se asignaron a recibir 2 años de quimioterapia con dasatinib.
- La tasa de SSC a 5 años fue del 54,6 %, y la tasa de SG a 5 años fue del 91,5 %, lo que indica una tasa alta de posibilidad de recuperación después de la recaída.
- En los pacientes con LLA de riesgo estándar, la tasa de SSC a 5 años fue del 52,1 %, y la tasa de SG fue del 82,3 %.
- En los pacientes con LLA de riesgo alto, la tasa de SSC a 5 años fue del 56,8 %, y la tasa de SG fue del 78,2 %.
- La tasa de SSC a 3 años en el ensayo AALL1122 trial (65,5 %) no fue inferior desde el punto de vista estadístico a la del ensayo EsPhALL de 2010 (59,1 %), en donde los pacientes recibieron imatinib combinado con la misma quimioterapia de base. Sin embargo, menos pacientes del ensayo AALL1122 se sometieron a TCMH en la primera RC, en comparación con el ensayo EsPhALL de 2010 (14 % vs. 38 %). Asímismo, menos pacientes en el ensayo AALL1122 recibieron radioterapia craneal (15 % vs. 100 %).
- La frecuencia de infecciones fue alta (sepsis de grado 3 o superior, 34 %; infección micótica de grado 3 superior, 14 %), y la tasa de mortalidad relacionada con el tratamiento fue del 8 % (8 % en quienes recibieron quimioterapia con dasatinib, 13 % en quienes recibieron un TCMH en el momento de la primera RC).
- La ERM en este ensayo se evaluó mediante 3 métodos: PCR de IG/TCR, citometría de flujo y PCR de BCR::ABL1. El grado de concordancia entre las pruebas fue más alto para la PCR de IG/TCR y la citometría de flujo, mientras que los resultados para la PCR de BCR::ABL1 fueron, en general, más discordantes. El ensayo de PCR de BCR::ABL1 fue el que más frecuentemente persistió positivo mientras que las otras pruebas rendían resultados indetectables. El desenlace de los pacientes con persistencia de resultados de positividad en la PCR de BCR::ABL1, pero que tenían resultados negativos para ERM en otras 2 pruebas, fue similar al desenlace de los pacientes con resultados no detectables en las 3 pruebas.
- En el ensayo ALL-2015 del Chinese Children’s Cancer Group 189 pacientes se asignaron al azar para recibir dasatinib o imatinib en combinación con un régimen multifarmacológico de acuerdo a los protocolos del SJCRH ALL. En este este ensayo, el dasatinib se administró en dosis más altas (80 mg/m2 en lugar de 60 mg/m2) y el imatinib se administró en dosis más bajas (300 mg/m2 en lugar de 340 mg/m2) que las que se habían administrado en los ensayos pediátricos previos de LLA BCR::ABL1 realizados por el COG y el EsPhALL.[Nivel de evidencia A1]
- Al cabo de una mediana de seguimiento de 26,4 meses, la SSE a 4 años y las tasas de SG fueron del 71 % y del 88,4 %, respectivamente, en los pacientes que se asignaron al azar a recibir dasatinib, en comparación con el 48,9 % y el 69,2 %, respectivamente, en los pacientes que se asignaron al azar a recibir imatinib.
- La toxicidad fue similar en ambos grupos de tratamiento.
- Se debe ser cauto al interpretar estos resultados debido a que la mediana de seguimiento fue corta y porque los desenlaces de los pacientes que recibieron imatinib en este ensayo fueron inferiores a los resultados de los pacientes tratados con imatinib notificados en ensayos anteriores.
Opciones de tratamiento en evaluación clínica para la leucemia linfoblástica aguda BCR::ABL1
La información en inglés sobre los ensayos clínicos patrocinados por el NCI se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de un ensayo clínico nacional o institucional en curso:
- AALL1631 (NCT03007147) (Imatinib Mesylate and Combination Chemotherapy in Treating Patients with Newly Diagnosed BCR::ABL1 ALL): AALL1631 es un protocolo colaborativo internacional dirigido por el COG y el grupo europeo EsPhALL. Los pacientes con LLA BCR::ABL1 ingresan al ensayo el día 15 de la inducción IA y en ese momento empiezan a recibir imatinib todos los días. Después de la fase de inducción IB (semanas 10–12), se evalúa la ERM mediante PCR para la inmunoglobulina H y el receptor de célula T (IgH-TCR), de manera que los pacientes se clasifican como de riesgo estándar (ERM <0,05 %) o riesgo alto (ERM >0,05 %). Los pacientes de riesgo estándar se asignan al azar a recibir uno de los siguientes regímenes de quimioterapia citotóxica de base:
- El tratamiento de base EsPhALL utilizado en los protocolos anteriores de EsPhALL y COG AALL1122; o
- Un régimen menos intensivo similar a los de uso común en los ensayos del COG para los pacientes con LLA de células B de riesgo alto que no presentan BCR::ABL1.
Los pacientes de riesgo estándar de ambos grupos continuarán recibiendo imatinib hasta que completen toda la quimioterapia prevista (2 años de tratamiento). El objetivo de la aleatorización al grupo de riesgo estándar es determinar si la quimioterapia de base menos intensiva se relaciona con una SSE similar, pero con tasas más bajas de toxicidad del tratamiento en comparación con la terapia estándar (quimioterapia de base de EsPhALL).
Los pacientes de riesgo alto (cerca del 15 % al 20 % de los pacientes) se someterán a un TCMH después de completar los 3 bloques de quimioterapia de consolidación. Se reiniciará el imatinib después del TCMH y se administrará desde el día +56 hasta el día +365 a fin de probar la viabilidad de la administración de este fármaco después del TCMH y describir el desenlace de los pacientes que reciben dicho tratamiento.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
References
- Hunger SP, Lu X, Devidas M, et al.: Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol 30 (14): 1663-9, 2012.
- Burns MA, Place AE, Stevenson KE, et al.: Identification of prognostic factors in childhood T-cell acute lymphoblastic leukemia: Results from DFCI ALL Consortium Protocols 05-001 and 11-001. Pediatr Blood Cancer 68 (1): e28719, 2021.
- Winter SS, Dunsmore KP, Devidas M, et al.: Improved Survival for Children and Young Adults With T-Lineage Acute Lymphoblastic Leukemia: Results From the Children's Oncology Group AALL0434 Methotrexate Randomization. J Clin Oncol 36 (29): 2926-2934, 2018.
- LeClerc JM, Billett AL, Gelber RD, et al.: Treatment of childhood acute lymphoblastic leukemia: results of Dana-Farber ALL Consortium Protocol 87-01. J Clin Oncol 20 (1): 237-46, 2002.
- Asselin BL, Devidas M, Wang C, et al.: Effectiveness of high-dose methotrexate in T-cell lymphoblastic leukemia and advanced-stage lymphoblastic lymphoma: a randomized study by the Children's Oncology Group (POG 9404). Blood 118 (4): 874-83, 2011.
- Silverman LB, Stevenson KE, O'Brien JE, et al.: Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia 24 (2): 320-34, 2010.
- Asselin BL, Devidas M, Chen L, et al.: Cardioprotection and Safety of Dexrazoxane in Patients Treated for Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma: A Report of the Children's Oncology Group Randomized Trial Pediatric Oncology Group 9404. J Clin Oncol 34 (8): 854-62, 2016.
- Chow EJ, Asselin BL, Schwartz CL, et al.: Late Mortality After Dexrazoxane Treatment: A Report From the Children's Oncology Group. J Clin Oncol 33 (24): 2639-45, 2015.
- Seibel NL, Asselin BL, Nachman JB, et al.: Treatment of high risk T-cell acute lymphoblastic leukemia (T-ALL): comparison of recent experience of the Children’s Cancer Group (CCG) and Pediatric Oncology Group (POG). [Abstract] Blood 104 (11): A-681, 2004.
- Seibel NL, Steinherz PG, Sather HN, et al.: Early postinduction intensification therapy improves survival for children and adolescents with high-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 111 (5): 2548-55, 2008.
- Hastings C, Gaynon PS, Nachman JB, et al.: Increased post-induction intensification improves outcome in children and adolescents with a markedly elevated white blood cell count (≥200 × 10(9) /l) with T cell acute lymphoblastic leukaemia but not B cell disease: a report from the Children's Oncology Group. Br J Haematol 168 (4): 533-46, 2015.
- Matloub Y, Stork L, Asselin B, et al.: Outcome of Children with Standard-Risk T-Lineage Acute Lymphoblastic Leukemia--Comparison among Different Treatment Strategies. Pediatr Blood Cancer 63 (2): 255-61, 2016.
- Berg SL, Blaney SM, Devidas M, et al.: Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children's Oncology Group. J Clin Oncol 23 (15): 3376-82, 2005.
- Kurtzberg J, Ernst TJ, Keating MJ, et al.: Phase I study of 506U78 administered on a consecutive 5-day schedule in children and adults with refractory hematologic malignancies. J Clin Oncol 23 (15): 3396-403, 2005.
- Winter SS, Dunsmore KP, Devidas M, et al.: Safe integration of nelarabine into intensive chemotherapy in newly diagnosed T-cell acute lymphoblastic leukemia: Children's Oncology Group Study AALL0434. Pediatr Blood Cancer 62 (7): 1176-83, 2015.
- Dunsmore KP, Devidas M, Linda SB, et al.: Pilot study of nelarabine in combination with intensive chemotherapy in high-risk T-cell acute lymphoblastic leukemia: a report from the Children's Oncology Group. J Clin Oncol 30 (22): 2753-9, 2012.
- Dunsmore KP, Winter SS, Devidas M, et al.: Children's Oncology Group AALL0434: A Phase III Randomized Clinical Trial Testing Nelarabine in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia. J Clin Oncol 38 (28): 3282-3293, 2020.
- Gossai NP, Devidas M, Chen Z, et al.: Central nervous system status is prognostic in T-cell acute lymphoblastic leukemia: a Children's Oncology Group report. Blood 141 (15): 1802-1811, 2023.
- Teachey DT, Devidas M, Wood BL, et al.: Children's Oncology Group Trial AALL1231: A Phase III Clinical Trial Testing Bortezomib in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia and Lymphoma. J Clin Oncol 40 (19): 2106-2118, 2022.
- Silverman LB: Acute lymphoblastic leukemia in infancy. Pediatr Blood Cancer 49 (7 Suppl): 1070-3, 2007.
- Hilden JM, Dinndorf PA, Meerbaum SO, et al.: Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children's Oncology Group. Blood 108 (2): 441-51, 2006.
- Pieters R, Schrappe M, De Lorenzo P, et al.: A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370 (9583): 240-50, 2007.
- Dreyer ZE, Hilden JM, Jones TL, et al.: Intensified chemotherapy without SCT in infant ALL: results from COG P9407 (Cohort 3). Pediatr Blood Cancer 62 (3): 419-26, 2015.
- Tomizawa D, Miyamura T, Imamura T, et al.: A risk-stratified therapy for infants with acute lymphoblastic leukemia: a report from the JPLSG MLL-10 trial. Blood 136 (16): 1813-1823, 2020.
- van der Linden MH, Valsecchi MG, De Lorenzo P, et al.: Outcome of congenital acute lymphoblastic leukemia treated on the Interfant-99 protocol. Blood 114 (18): 3764-8, 2009.
- Tomizawa D, Koh K, Sato T, et al.: Outcome of risk-based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: a final report of two consecutive studies, MLL96 and MLL98, of the Japan Infant Leukemia Study Group. Leukemia 21 (11): 2258-63, 2007.
- Brown PA, Kairalla JA, Hilden JM, et al.: FLT3 inhibitor lestaurtinib plus chemotherapy for newly diagnosed KMT2A-rearranged infant acute lymphoblastic leukemia: Children's Oncology Group trial AALL0631. Leukemia 35 (5): 1279-1290, 2021.
- Van der Velden VH, Corral L, Valsecchi MG, et al.: Prognostic significance of minimal residual disease in infants with acute lymphoblastic leukemia treated within the Interfant-99 protocol. Leukemia 23 (6): 1073-9, 2009.
- Pieters R, De Lorenzo P, Ancliffe P, et al.: Outcome of Infants Younger Than 1 Year With Acute Lymphoblastic Leukemia Treated With the Interfant-06 Protocol: Results From an International Phase III Randomized Study. J Clin Oncol 37 (25): 2246-2256, 2019.
- Popov A, Tsaur G, Permikin Z, et al.: Incidence and prognostic value of central nervous system involvement in infants with B-cell precursor acute lymphoblastic leukemia treated according to the MLL-Baby protocol. Pediatr Blood Cancer 69 (9): e29860, 2022.
- Ibrahimova A, Winestone LE, Miller TP, et al.: Presentation acuity, induction mortality, and resource utilization in infants with acute leukemia. Pediatr Blood Cancer 68 (7): e28940, 2021.
- Salzer WL, Jones TL, Devidas M, et al.: Decreased induction morbidity and mortality following modification to induction therapy in infants with acute lymphoblastic leukemia enrolled on AALL0631: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (3): 414-8, 2015.
- Stutterheim J, van der Sluis IM, de Lorenzo P, et al.: Clinical Implications of Minimal Residual Disease Detection in Infants With KMT2A-Rearranged Acute Lymphoblastic Leukemia Treated on the Interfant-06 Protocol. J Clin Oncol 39 (6): 652-662, 2021.
- van der Sluis IM, de Lorenzo P, Kotecha RS, et al.: Blinatumomab Added to Chemotherapy in Infant Lymphoblastic Leukemia. N Engl J Med 388 (17): 1572-1581, 2023.
- Kosaka Y, Koh K, Kinukawa N, et al.: Infant acute lymphoblastic leukemia with MLL gene rearrangements: outcome following intensive chemotherapy and hematopoietic stem cell transplantation. Blood 104 (12): 3527-34, 2004.
- Dreyer ZE, Dinndorf PA, Camitta B, et al.: Analysis of the role of hematopoietic stem-cell transplantation in infants with acute lymphoblastic leukemia in first remission and MLL gene rearrangements: a report from the Children's Oncology Group. J Clin Oncol 29 (2): 214-22, 2011.
- Mann G, Attarbaschi A, Schrappe M, et al.: Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-lineage-leukemia (MLL)-rearranged acute lymphoblastic leukemia: results from the Interfant-99 Study. Blood 116 (15): 2644-50, 2010.
- Kato M, Hasegawa D, Koh K, et al.: Allogeneic haematopoietic stem cell transplantation for infant acute lymphoblastic leukaemia with KMT2A (MLL) rearrangements: a retrospective study from the paediatric acute lymphoblastic leukaemia working group of the Japan Society for Haematopoietic Cell Transplantation. Br J Haematol 168 (4): 564-70, 2015.
- Stutterheim J, de Lorenzo P, van der Sluis IM, et al.: Minimal residual disease and outcome characteristics in infant KMT2A-germline acute lymphoblastic leukaemia treated on the Interfant-06 protocol. Eur J Cancer 160: 72-79, 2022.
- Guest EM, Kairalla JA, Hilden JM, et al.: Outstanding outcomes in infants with KMT2A-germline acute lymphoblastic leukemia treated with chemotherapy alone: results of the Children's Oncology Group AALL0631 trial. Haematologica 107 (5): 1205-1208, 2022.
- Nachman J: Clinical characteristics, biologic features and outcome for young adult patients with acute lymphoblastic leukaemia. Br J Haematol 130 (2): 166-73, 2005.
- Pui CH, Pei D, Campana D, et al.: Improved prognosis for older adolescents with acute lymphoblastic leukemia. J Clin Oncol 29 (4): 386-91, 2011.
- Nachman JB, La MK, Hunger SP, et al.: Young adults with acute lymphoblastic leukemia have an excellent outcome with chemotherapy alone and benefit from intensive postinduction treatment: a report from the children's oncology group. J Clin Oncol 27 (31): 5189-94, 2009.
- Pichler H, Reismüller B, Steiner M, et al.: The inferior prognosis of adolescents with acute lymphoblastic leukaemia (ALL) is caused by a higher rate of treatment-related mortality and not an increased relapse rate--a population-based analysis of 25 years of the Austrian ALL-BFM (Berlin-Frankfurt-Münster) Study Group. Br J Haematol 161 (4): 556-65, 2013.
- Burke MJ, Gossai N, Wagner JE, et al.: Survival differences between adolescents/young adults and children with B precursor acute lymphoblastic leukemia after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 19 (1): 138-42, 2013.
- Bhatia S, Landier W, Shangguan M, et al.: Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the children's oncology group. J Clin Oncol 30 (17): 2094-101, 2012.
- Stock W, La M, Sanford B, et al.: What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children's Cancer Group and Cancer and Leukemia Group B studies. Blood 112 (5): 1646-54, 2008.
- Ramanujachar R, Richards S, Hann I, et al.: Adolescents with acute lymphoblastic leukaemia: emerging from the shadow of paediatric and adult treatment protocols. Pediatr Blood Cancer 47 (6): 748-56, 2006.
- Barry E, DeAngelo DJ, Neuberg D, et al.: Favorable outcome for adolescents with acute lymphoblastic leukemia treated on Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium Protocols. J Clin Oncol 25 (7): 813-9, 2007.
- Ramanujachar R, Richards S, Hann I, et al.: Adolescents with acute lymphoblastic leukaemia: outcome on UK national paediatric (ALL97) and adult (UKALLXII/E2993) trials. Pediatr Blood Cancer 48 (3): 254-61, 2007.
- Ram R, Wolach O, Vidal L, et al.: Adolescents and young adults with acute lymphoblastic leukemia have a better outcome when treated with pediatric-inspired regimens: systematic review and meta-analysis. Am J Hematol 87 (5): 472-8, 2012.
- Boissel N, Auclerc MF, Lhéritier V, et al.: Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-93 and LALA-94 trials. J Clin Oncol 21 (5): 774-80, 2003.
- Huguet F, Leguay T, Raffoux E, et al.: Pediatric-inspired therapy in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: the GRAALL-2003 study. J Clin Oncol 27 (6): 911-8, 2009.
- DeAngelo DJ, Stevenson KE, Dahlberg SE, et al.: Long-term outcome of a pediatric-inspired regimen used for adults aged 18-50 years with newly diagnosed acute lymphoblastic leukemia. Leukemia 29 (3): 526-34, 2015.
- Advani AS, Larsen E, Laumann K, et al.: Comparison of CALGB 10403 (Alliance) and COG AALL0232 toxicity results in young adults with acute lymphoblastic leukemia. Blood Adv 5 (2): 504-512, 2021.
- Ribera JM, Oriol A, Sanz MA, et al.: Comparison of the results of the treatment of adolescents and young adults with standard-risk acute lymphoblastic leukemia with the Programa Español de Tratamiento en Hematología pediatric-based protocol ALL-96. J Clin Oncol 26 (11): 1843-9, 2008.
- Stock W, Luger SM, Advani AS, et al.: A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: results of CALGB 10403. Blood 133 (14): 1548-1559, 2019.
- Wieduwilt MJ, Stock W, Advani A, et al.: Superior survival with pediatric-style chemotherapy compared to myeloablative allogeneic hematopoietic cell transplantation in older adolescents and young adults with Ph-negative acute lymphoblastic leukemia in first complete remission: analysis from CALGB 10403 and the CIBMTR. Leukemia 35 (7): 2076-2085, 2021.
- Gupta S, Pole JD, Baxter NN, et al.: The effect of adopting pediatric protocols in adolescents and young adults with acute lymphoblastic leukemia in pediatric vs adult centers: An IMPACT Cohort study. Cancer Med 8 (5): 2095-2103, 2019.
- Testi AM, Valsecchi MG, Conter V, et al.: Difference in outcome of adolescents with acute lymphoblastic leukemia (ALL) enrolled in pediatric (AIEOP) and adult (GIMEMA) protocols. [Abstract] Blood 104: A-1954, 2004.
- de Bont JM, van der Holt B, Dekker AW, et al.: [Adolescents with acute lymphatic leukaemia achieve significantly better results when treated following Dutch paediatric oncology protocols than with adult protocols]. Ned Tijdschr Geneeskd 149 (8): 400-6, 2005.
- Hallböök H, Gustafsson G, Smedmyr B, et al.: Treatment outcome in young adults and children >10 years of age with acute lymphoblastic leukemia in Sweden: a comparison between a pediatric protocol and an adult protocol. Cancer 107 (7): 1551-61, 2006.
- Hough R, Rowntree C, Goulden N, et al.: Efficacy and toxicity of a paediatric protocol in teenagers and young adults with Philadelphia chromosome negative acute lymphoblastic leukaemia: results from UKALL 2003. Br J Haematol 172 (3): 439-51, 2016.
- Mattano LA, Devidas M, Nachman JB, et al.: Effect of alternate-week versus continuous dexamethasone scheduling on the risk of osteonecrosis in paediatric patients with acute lymphoblastic leukaemia: results from the CCG-1961 randomised cohort trial. Lancet Oncol 13 (9): 906-15, 2012.
- Mogensen SS, Harila-Saari A, Mäkitie O, et al.: Comparing osteonecrosis clinical phenotype, timing, and risk factors in children and young adults treated for acute lymphoblastic leukemia. Pediatr Blood Cancer 65 (10): e27300, 2018.
- Larsen EC, Devidas M, Chen S, et al.: Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults With High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group Study AALL0232. J Clin Oncol 34 (20): 2380-8, 2016.
- Zeller B, Gustafsson G, Forestier E, et al.: Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br J Haematol 128 (6): 797-804, 2005.
- Arico M, Ziino O, Valsecchi MG, et al.: Acute lymphoblastic leukemia and Down syndrome: presenting features and treatment outcome in the experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP). Cancer 113 (3): 515-21, 2008.
- Maloney KW, Carroll WL, Carroll AJ, et al.: Down syndrome childhood acute lymphoblastic leukemia has a unique spectrum of sentinel cytogenetic lesions that influences treatment outcome: a report from the Children's Oncology Group. Blood 116 (7): 1045-50, 2010.
- de Graaf G, Buckley F, Skotko BG: Estimation of the number of people with Down syndrome in the United States. Genet Med 19 (4): 439-447, 2017.
- Chessells JM, Harrison G, Richards SM, et al.: Down's syndrome and acute lymphoblastic leukaemia: clinical features and response to treatment. Arch Dis Child 85 (4): 321-5, 2001.
- Buitenkamp TD, Izraeli S, Zimmermann M, et al.: Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood 123 (1): 70-7, 2014.
- Mullighan CG, Collins-Underwood JR, Phillips LA, et al.: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243-6, 2009.
- Bercovich D, Ganmore I, Scott LM, et al.: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484-92, 2008.
- Gaikwad A, Rye CL, Devidas M, et al.: Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol 144 (6): 930-2, 2009.
- Kearney L, Gonzalez De Castro D, Yeung J, et al.: Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood 113 (3): 646-8, 2009.
- Buitenkamp TD, Pieters R, Gallimore NE, et al.: Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia 26 (10): 2204-11, 2012.
- Hanada I, Terui K, Ikeda F, et al.: Gene alterations involving the CRLF2-JAK pathway and recurrent gene deletions in Down syndrome-associated acute lymphoblastic leukemia in Japan. Genes Chromosomes Cancer 53 (11): 902-10, 2014.
- Bassal M, La MK, Whitlock JA, et al.: Lymphoblast biology and outcome among children with Down syndrome and ALL treated on CCG-1952. Pediatr Blood Cancer 44 (1): 21-8, 2005.
- Whitlock JA, Sather HN, Gaynon P, et al.: Clinical characteristics and outcome of children with Down syndrome and acute lymphoblastic leukemia: a Children's Cancer Group study. Blood 106 (13): 4043-9, 2005.
- Lundin C, Forestier E, Klarskov Andersen M, et al.: Clinical and genetic features of pediatric acute lymphoblastic leukemia in Down syndrome in the Nordic countries. J Hematol Oncol 7 (1): 32, 2014.
- Athale UH, Puligandla M, Stevenson KE, et al.: Outcome of children and adolescents with Down syndrome treated on Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium protocols 00-001 and 05-001. Pediatr Blood Cancer 65 (10): e27256, 2018.
- Matloub Y, Rabin KR, Ji L, et al.: Excellent long-term survival of children with Down syndrome and standard-risk ALL: a report from the Children's Oncology Group. Blood Adv 3 (11): 1647-1656, 2019.
- Aricò M, Valsecchi MG, Camitta B, et al.: Outcome of treatment in children with Philadelphia chromosome-positive acute lymphoblastic leukemia. N Engl J Med 342 (14): 998-1006, 2000.
- Aricò M, Schrappe M, Hunger SP, et al.: Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol 28 (31): 4755-61, 2010.
- Zuna J, Hovorkova L, Krotka J, et al.: Minimal residual disease in BCR::ABL1-positive acute lymphoblastic leukemia: different significance in typical ALL and in CML-like disease. Leukemia 36 (12): 2793-2801, 2022.
- Short NJ, Jabbour E, Macaron W, et al.: Ultrasensitive NGS MRD assessment in Ph+ ALL: Prognostic impact and correlation with RT-PCR for BCR::ABL1. Am J Hematol 98 (8): 1196-1203, 2023.
- Hunger SP, Tran TH, Saha V, et al.: Dasatinib with intensive chemotherapy in de novo paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (CA180-372/COG AALL1122): a single-arm, multicentre, phase 2 trial. Lancet Haematol 10 (7): e510-e520, 2023.
- Hovorkova L, Zaliova M, Venn NC, et al.: Monitoring of childhood ALL using BCR-ABL1 genomic breakpoints identifies a subgroup with CML-like biology. Blood 129 (20): 2771-2781, 2017.
- Duffield AS, Mullighan CG, Borowitz MJ: International Consensus Classification of acute lymphoblastic leukemia/lymphoma. Virchows Arch 482 (1): 11-26, 2023.
- Champagne MA, Capdeville R, Krailo M, et al.: Imatinib mesylate (STI571) for treatment of children with Philadelphia chromosome-positive leukemia: results from a Children's Oncology Group phase 1 study. Blood 104 (9): 2655-60, 2004.
- Ottmann OG, Druker BJ, Sawyers CL, et al.: A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood 100 (6): 1965-71, 2002.
- Thomas DA, Faderl S, Cortes J, et al.: Treatment of Philadelphia chromosome-positive acute lymphocytic leukemia with hyper-CVAD and imatinib mesylate. Blood 103 (12): 4396-407, 2004.
- Yanada M, Takeuchi J, Sugiura I, et al.: High complete remission rate and promising outcome by combination of imatinib and chemotherapy for newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia: a phase II study by the Japan Adult Leukemia Study Group. J Clin Oncol 24 (3): 460-6, 2006.
- Schultz KR, Bowman WP, Aledo A, et al.: Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 27 (31): 5175-81, 2009.
- Burke MJ, Trotz B, Luo X, et al.: Allo-hematopoietic cell transplantation for Ph chromosome-positive ALL: impact of imatinib on relapse and survival. Bone Marrow Transplant 43 (2): 107-13, 2009.
- Lee S, Kim YJ, Min CK, et al.: The effect of first-line imatinib interim therapy on the outcome of allogeneic stem cell transplantation in adults with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood 105 (9): 3449-57, 2005.
- de Labarthe A, Rousselot P, Huguet-Rigal F, et al.: Imatinib combined with induction or consolidation chemotherapy in patients with de novo Philadelphia chromosome-positive acute lymphoblastic leukemia: results of the GRAAPH-2003 study. Blood 109 (4): 1408-13, 2007.
- Rives S, Estella J, Gómez P, et al.: Intermediate dose of imatinib in combination with chemotherapy followed by allogeneic stem cell transplantation improves early outcome in paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (ALL): results of the Spanish Cooperative Group SHOP studies ALL-94, ALL-99 and ALL-2005. Br J Haematol 154 (5): 600-11, 2011.
- Schultz KR, Carroll A, Heerema NA, et al.: Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's Oncology Group study AALL0031. Leukemia 28 (7): 1467-71, 2014.
- Biondi A, Schrappe M, De Lorenzo P, et al.: Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol 13 (9): 936-45, 2012.
- Porkka K, Koskenvesa P, Lundán T, et al.: Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood 112 (4): 1005-12, 2008.
- Zwaan CM, Rizzari C, Mechinaud F, et al.: Dasatinib in children and adolescents with relapsed or refractory leukemia: results of the CA180-018 phase I dose-escalation study of the Innovative Therapies for Children with Cancer Consortium. J Clin Oncol 31 (19): 2460-8, 2013.
- Slayton WB, Schultz KR, Kairalla JA, et al.: Dasatinib Plus Intensive Chemotherapy in Children, Adolescents, and Young Adults With Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: Results of Children's Oncology Group Trial AALL0622. J Clin Oncol 36 (22): 2306-2314, 2018.
- Biondi A, Cario G, De Lorenzo P, et al.: Long-term follow up of pediatric Philadelphia positive acute lymphoblastic leukemia treated with the EsPhALL2004 study: high white blood cell count at diagnosis is the strongest prognostic factor. Haematologica 104 (1): e13-e16, 2019.
- Biondi A, Gandemer V, De Lorenzo P, et al.: Imatinib treatment of paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (EsPhALL2010): a prospective, intergroup, open-label, single-arm clinical trial. Lancet Haematol 5 (12): e641-e652, 2018.
- Shen S, Chen X, Cai J, et al.: Effect of Dasatinib vs Imatinib in the Treatment of Pediatric Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA Oncol 6 (3): 358-366, 2020.
Tratamiento de la leucemia linfoblástica aguda infantil en recaída
Factores pronósticos después de la primera recaída de la leucemia linfoblástica aguda infantil
El pronóstico de un niño con leucemia linfoblástica aguda (LLA) en recaída depende de varios factores.; [Nivel de evidencia C2]
Después de la primera recaída de la LLA infantil se utilizan los siguientes dos factores de riesgo importantes para determinar el pronóstico y el abordaje de tratamiento:
- Sitio de la recaída.
- Tiempo desde el diagnóstico hasta la recaída.
Otros factores pronósticos son los siguientes:
- Características del paciente (por ejemplo, edad y recuento de blastocitos periféricos en el momento de la recaída).
- Clasificación del grupo de riesgo en el momento del diagnóstico inicial.
- Respuesta a la terapia de reinducción.
- Alteraciones citogenéticas o genómicas.
- Inmunofenotipo.
Sitio de la recaída
Los pacientes con una recaída extramedular aislada por lo general tienen una evolución más favorable que los pacientes con recaídas en la médula ósea. En algunos estudios, los pacientes con recaídas medular y extramedular combinadas tuvieron un mejor pronóstico que aquellos con recaída medular. Sin embargo, este hallazgo no se confirmó en otros estudios.
Tiempo desde el diagnóstico hasta la recaída
En los pacientes con LLA-B recidivante, las recaídas tempranas y las recaídas medulares indican una evolución más desfavorable que las recaídas tardías y las recaídas extramedulares aisladas. Por ejemplo, las tasas de supervivencia oscilan entre menos de un 20 %, en los pacientes con recaídas medulares que surgen durante los 18 meses que le siguen al diagnóstico, hasta un 60 %, en aquellos cuyas recaídas se presentan después de 36 meses del diagnóstico.
En los pacientes con recaídas aisladas en el sistema nervioso central (SNC), las tasas de supervivencia general (SG) son del 40 % al 50 % cuando la recaída es temprana (<18 meses a partir del diagnóstico) y del 75 % al 80 % cuando la recaída es tardía (>18 meses a partir del diagnóstico). No se ha comprobado que el resultado mejore cuando se establece vigilancia frecuente (recuentos sanguíneos completos o pruebas de médula ósea) para la detección temprana de la recaída en los pacientes que no reciben tratamiento.
Características de las pacientes
Se notificó que una edad de 10 años o más en el momento del diagnóstico es un factor pronóstico independiente de un desenlace precario. En un estudio del Children’s Oncology Group (COG) se observó además que, aunque los pacientes de 10 a 15 años en el momento del diagnóstico inicial exhibían una evolución más desfavorable que los pacientes de 1 a 9 años (tasa de supervivencia posterior a la recaía a 3 años, 35 vs. 48 %), la evolución de los mayores de 15 años fue mucho más desfavorable (tasa de SG a 3 años,15 %; P = 0,001).
Para los pacientes con LLA-B diagnosticada a los 18 años de edad o antes y que tuvieron una recaída tardía, la edad no fue un factor pronóstico importante del desenlace subsiguiente cuando se hizo un análisis por cuartiles. Sin embargo, el desenlace de los pacientes de 18 años o más en el momento de la recaída fue significativamente inferior al desenlace de los pacientes que presentaron una recaída a una edad menor a los 18 años (39,5 vs. 68,7 %; P = 0,0001).
El grupo Berlin-Frankfurt-Münster (BFM) también notificó que un recuento de blastocitos periféricos elevado (>10 000/μl) en el momento de la recaída se relacionó con desenlaces inferiores en los pacientes con recaídas medulares tardías.
Los niños con síndrome de Down y recaída de LLA por lo general presentaron desenlaces inferiores debido al aumento de muertes durante la inducción, la mortalidad relacionada con el tratamiento y la recaída.
- El grupo BFM demostró que, desde el año 2000, las mejoras en los cuidados médicos de apoyo redujeron la mortalidad relacionada con el tratamiento de niños con síndrome de Down; sin embargo, el riesgo de recaída permanece alto.
- Esta conclusión se basó en un análisis de los datos del Center for International Blood and Marrow Transplant Research (CIBMTR) en 27 pacientes con síndrome de Down y LLA sometidos a un trasplante de células madre hematopoyéticas (TCMH) entre 2000 y 2009. Los investigadores observaron que al usar las prácticas de trasplante vigentes se obtenían tasas dentro del intervalo esperado de recuperación hematopoyética, incidencia de enfermedad de injerto contra huésped (EICH) y mortalidad relacionada con el trasplante, en comparación con los pacientes de LLA sin síndrome de Down. No obstante, la recaída fue más alta que la esperada (>50 %) y fue la causa principal de fracaso del tratamiento, lo que produjo una supervivencia precaria (tasa de supervivencia sin enfermedad [SSE] a 3 años, 24 %).[Nivel de evidencia C1]
Clasificación del grupo de riesgo en el momento del diagnóstico inicial
El COG informó que la clasificación del grupo de riesgo en el diagnóstico inicial fue importante para el pronóstico después de la recaída. Los pacientes que cumplieron con los criterios de riesgo estándar del Instituto Nacional del Cáncer (NCI) en el momento del diagnóstico inicial evolucionaron de forma más favorable después de la recaída que los pacientes de riesgo alto del NCI.
Respuesta a la terapia de reinducción
Los pacientes con recaídas medulares y enfermedad morfológica persistente al final del primer mes de terapia de reinducción tienen un pronóstico muy precario, incluso si luego logran una segunda remisión completa (RC).[Nivel de evidencia B4]; [Nivel de evidencia C1] En varios estudios se demostró que los índices de enfermedad residual mínima (ERM) después de la segunda RC tienen valor pronóstico en la LLA en recaída.; [Nivel de evidencia C2] Los índices altos de ERM al final de la reinducción y en puntos de referencia posteriores se correlacionaron con un riesgo muy alto de recaída posterior.
Alteraciones citogenéticas o genómicas
Mediante secuenciación génica se han identificado cambios en los perfiles de variantes desde el diagnóstico hasta el momento de la recaída. Las fusiones génicas oncógenas (por ejemplo, TCF3::PBX1, ETV6::RUNX1) casi siempre se observan entre el momento del diagnóstico inicial y la recaída; las variantes de un solo nucleótido y las variaciones en el número de copias a veces se observan en el momento del diagnóstico, pero no en el momento de la recaída y viceversa. Por ejemplo, las variantes de la familia de genes RAS son comunes en el momento del diagnóstico y de la recaída, pero el tipo de variante específica en la familia de genes RAS quizás cambie desde el diagnóstico hasta la recaída a medida que subclones leucémicos específicos surgen o desaparecen durante el curso del tratamiento. Por el contrario, se han observado variantes de NT5C2 (un gen que participa en el metabolismo de los nucleótidos) relacionadas de manera específica con la recaída hasta en el 45 % de los casos de LLA que exhiben recaída temprana.
Las alteraciones de TP53 (variantes o alteraciones en el número de copias) se observan en cerca del 10 % de los pacientes con LLA en la primera recaída y se relacionan con un aumento de la probabilidad de leucemia persistente después de la reinducción inicial y tasas de supervivencia sin complicaciones (SSC) precarias. En un estudio, cerca de la mitad de las alteraciones en TP53 se observaron en el momento del diagnóstico inicial y el resto se observaron por primera vez en el momento de la recaída.
También se notificó una asociación entre la presencia de deleciones de IKZF1 y un pronóstico precario para los pacientes con LLA-B en la primera recaída medular. Sin embargo, en un estudio del BFM de pacientes con LLA-B que tuvieron una primera recaída medular tardía, las deleciones de IKZF1 no tuvieron importancia pronóstica.
Las variantes de la vía RAS (KRAS, NRAS, FLT3 y PTPN11) son comunes en el momento de la recaída en los pacientes con LLA-B y se encontraron en cerca del 40 % de los pacientes en el momento de la primera recaída en un estudio de 206 niños. De la misma forma que en el momento del diagnóstico, la frecuencia de las variantes de la vía RAS en el momento de la recaída difiere según el subtipo citogenético (por ejemplo, frecuencia elevada en casos de hiperdiploidía y frecuencia baja en casos con ETV6::RUNX1). La presencia de variantes de la vía RAS en el momento de la recaída se relacionó con una recaída temprana. No obstante, la presencia de variantes de la vía RAS en el momento de la recaída no fue un factor pronóstico independiente del desenlace.
Los pacientes con LLA positiva para ETV6::RUNX1 tienen un pronóstico relativamente favorable en la primera recaída, lo que se corresponde con el alto porcentaje de estos pacientes que recaen más de 36 meses después del diagnóstico.
- En el estudio ALL-REZ BFM 2002 (NCT00114348), se observó una tasa de SSC del 84 % (± 7 %) en los pacientes de LLA ETV6::RUNX1 y recaída en la médula ósea. En este estudio, la primera remisión duró por lo menos 6 meses después de finalizar el tratamiento primario en el 94 % de los pacientes con ETV6::RUNX1; en un análisis multivariante, el tiempo hasta la recaída (pero no la presencia de ETV6::RUNX1) fue un factor pronóstico independiente del desenlace.
- De modo parecido, la tasa de SG a 5 años fue del 81 % en los pacientes con ETV6::RUNX1 inscritos en el ensayo 93 del French Acute Lymphoblastic Leukaemia Study Group (FRALLE) que presentaron recaída en cualquier sitio más de 36 meses después del diagnóstico. La presencia de ETV6::RUNX1 se relacionó con un resultado de supervivencia favorable, en comparación con otros pacientes con recaídas tardías. Sin embargo, la tasa de SG a 3 años fue solo del 31 % en los pacientes con ETV6::RUNX1 que presentaron una recaída temprana (<36 meses).
Inmunofenotipo
El inmunofenotipo es un factor pronóstico importante en el momento de la recaída. Los pacientes con LLA-T que presentan una recaída medular (aislada o combinada) en cualquier momento durante el tratamiento o después del tratamiento tienen menos probabilidades de lograr una segunda remisión y una SSC a largo plazo que los pacientes con LLA-B.
Opciones de tratamiento estándar para la primera recaída en la médula ósea de la leucemia linfoblástica aguda infantil
Las opciones de tratamiento estándar para la primera recaída en la médula ósea son las siguientes:
- Quimioterapia de reinducción.
- Terapia de posreinducción para pacientes que logran una segunda remisión completa.
Quimioterapia de reinducción
El tratamiento inicial de la recaída consiste en terapia de reinducción para lograr una segunda RC. Cuando se usa un régimen de reinducción de 4 fármacos (similar al que se administra a los pacientes de riesgo alto con diagnóstico reciente) o un régimen alternativo que incluye dosis altas de metotrexato y citarabina, cerca del 85 % de los pacientes con recidiva en la médula ósea logra una segunda RC al final del primer mes de tratamiento.; [Nivel de evidencia B4] Los pacientes con recidivas medulares tempranas tienen una tasa más baja de segunda RC morfológica (alrededor del 70 %) que aquellos con recidivas medulares tardías (alrededor del 95 %).
Evidencia (quimioterapia de reinducción):
- En un estudio del COG se usaron 3 bloques de terapia de reinducción intensiva con una combinación inicial de 4 fármacos que incluyó doxorrubicina seguida de 2 bloques de consolidación intensiva antes del TCMH o la quimioterapia de continuación.
- Se logró la segunda remisión después del bloque 1 en el 68 % de los pacientes con recaída temprana (<36 meses desde el diagnóstico inicial) y en el 96 % de aquellos con recaída posterior.
- El uso de los bloques 2 y 3 redujo la ERM en 40 de 56 pacientes que presentaban ERM después del bloque 1.
- En un ensayo aleatorizado del Reino Unido de pacientes con LLA en primera recaída, se comparó la reinducción con una combinación de 4 fármacos con idarrubicina o mitoxantrona.[Nivel de evidencia A1]
- No hubo diferencia en las tasas de segunda RC ni en los índices de ERM al final de la reinducción en los 2 grupos del estudio.
- Se notificó una mejora significativa de la tasa de SG en el grupo de mitoxantrona (69 vs. 45 %, P = 0,007) debido a la disminución de recidivas después del trasplante.
Es necesario investigar más el posible beneficio de la mitoxantrona en los regímenes para la LLA en recaída.
- Los investigadores del grupo ALL-REZ BFM usaron un abordaje de reinducción de 6 fármacos que incluyó dosis altas de metotrexato.
- En una comparación aleatorizada de 1 g/m2 de metotrexato administrado en un plazo de 36 horas versus 5 g/m2 de metotrexato administrado en un plazo de 24 horas durante la reinducción, no se observó diferencia en el desenlace, pero la infusión de 36 horas se relacionó con una incidencia más alta de mucositis.
- Se notificó que la combinación de clofarabina, ciclofosfamida y etopósido induce la remisión en el 42 % al 56 % de los pacientes con enfermedad resistente al tratamiento o con recaídas múltiples.; [Nivel de evidencia B4]
- Se notificó que la combinación de bortezomib con vincristina, dexametasona, pegaspargasa y doxorrubicina induce una respuesta completa (con recuperación de plaquetas o sin esta) en el 70 % al 80 % de los pacientes con LLA-B que tienen recaídas múltiples.[Nivel de evidencia C1]; [Nivel de evidencia C3]
- Esta combinación (con prednisona en lugar de dexametasona) se probó en pacientes de LLA-B con una primer recaída antes de los 36 meses desde el diagnóstico inicial.
- La tasa de segunda RC fue del 68 %, lo cual no difiere significativamente de lo observado en un ensayo anterior en el que se usó la misma base de inducción sin bortezomib.
- En análisis de subgrupos, la adición de bortezomib a la base de reinducción de 4 fármacos no produjo tasas de segunda RC significativamente superiores en los pacientes con recaídas muy tempranas (<18 meses desde el diagnóstico) o recaídas tempranas (18–36 meses desde el diagnóstico) cuando se compararon con los controles históricos.
- Esta combinación (con prednisona en lugar de dexametasona) se probó en pacientes de LLA-B con una primer recaída antes de los 36 meses desde el diagnóstico inicial.
Los pacientes con LLA-T en recaída tienen tasas mucho más bajas de segunda RC que los pacientes con fenotipo de células B cuando reciben regímenes estándar de reinducción. En los niños con LLA-T que presentaron la primera recaída en la médula ósea, el tratamiento con monoterapia de nelarabina, un fármaco selectivo de células T, produjo tasas de respuesta de cerca del 50 %. En un ensayo de 28 pacientes pediátricos con LLA-T en recaída o resistente al tratamiento que se trataron con monoterapia de nelarabina, la tasa de respuesta general (RC y RC sin recuperación hematológica) fue del 39,3 %. La combinación de nelarabina, ciclofosfamida y etopósido también se ha usado en pacientes con LLA-T en recaída o resistente al tratamiento, con tasas de respuesta similares a las de la monoterapia de nelarabina.; [Nivel de evidencia C3] En un ensayo de fase I/II de esta combinación en pacientes con LLA-T y linfoma linfoblástico T en recaída o resistente al tratamiento, la mejor tasa de respuesta general (RC + RC con recuperación incompleta de plaquetas + respuesta parcial) fue del 38 % (8 de 21 pacientes). La cohorte de LLA-T tuvo una tasa de respuesta del 33 % (4 de 12 pacientes) y la cohorte de linfoma linfoblástico T tuvo una tasa de respuesta del 44 % (4 de 9 pacientes).[Nivel de evidencia C3]
En los pacientes con LLA-T en recaída también se evaluó el uso del inhibidor del proteosoma bortezomib (en combinación con quimioterapia). En un ensayo de fase II que realizó el COG, la combinación de bortezomib con vincristina, prednisona, pegaspargasa y doxorrubicina produjo una tasa de segunda RC del 68 % en los pacientes con LLA-T en primera remisión.
El fracaso de la reinducción es un factor de pronóstico precario, pero los intentos subsiguientes para lograr la remisión a veces son exitosos y llevan a supervivencia después de un TCMH, en especial, si la ERM es baja o indetectable. Para obtener más información sobre la estratificación del riesgo de ERM, consultar la sección Leucemia linfoblástica aguda de células B en recaída tardía. Tradicionalmente, los abordajes incluían el uso de combinaciones de fármacos diferentes a los usados en el primer intento de tratamiento. Estos regímenes a menudo incluían fármacos más nuevos en investigación en ensayos clínicos. Aunque la probabilidad de supervivencia disminuye en forma progresiva después de cada intento, por lo general, se realizan entre 2 y 4 intentos adicionales con mediciones de eficacia decrecientes después de cada uno. A raíz de que los estudios de células T con receptor de antígeno quimérico (CAR), blinatumomab e inotuzumab hayan revelado tasas altas de remisión en pacientes de LLA-B con múltiples recaídas y resistentes a la quimioterapia, se encuentran en marcha ensayos para evaluar estos fármacos después de la recaída inicial. Para obtener más información, consultar la sección Abordajes inmunoterapéuticos para la leucemia linfoblástica aguda en recaída o resistente al tratamiento.
Terapia de posreinducción en los pacientes que logran una segunda remisión completa
Leucemia linfoblástica aguda de células B en recaída temprana
En la mayoría de los estudios se notificó que, en los pacientes de LLA-B en recaída medular temprana, el trasplante alogénico de un donante fraterno con HLA idéntico o de un donante no emparentado compatible que se realiza en el momento de la segunda remisión conlleva una supervivencia sin leucemia (SSL) más alta que la de un abordaje quimioterapéutico. Sin embargo, incluso con el trasplante, la tasa de supervivencia para los pacientes con recaída medular temprana es inferior al 50 %. En un estudio se analizaron 278 pacientes con LLA-B en recaída temprana que recibieron tratamiento entre 2001 y 2013 en ensayos realizados por los grupos ALL-REZ BFM o UK-ALL. La tasa de supervivencia general fue del 32,6 %. Se observaron desenlaces más favorables en los pacientes con LLA-B en recaída temprana que tenían ERM baja al final de la reinducción, ERM baja antes del TCMH y en aquellos que presentaron EICH aguda. Para obtener más información, consultar la sección Trasplante de células madre hematopoyéticas en la primera recaída medular y en recaídas posteriores.
Después de la quimioterapia de reinducción inicial, se ha demostrado que el uso de blinatumomab en lugar de quimioterapia citotóxica intensiva como terapia de consolidación previa al TCMH se asocia con mejores resultados.
Evidencia (blinatumomab antes del TCMH en la LLA-B con recaída temprana):
- En un ensayo de fase III aleatorizado que se realizó entre 2015 y 2019, los pacientes con LLA-B en recaída temprana se asignaron al azar para recibir un curso de blinatumomab de 28 días o un ciclo de quimioterapia citotóxica intensiva como un tercer curso de consolidación antes de someterse al TCMH. En el estudio se esperaba inscribir a 202 pacientes, pero se interrumpió antes de tiempo (después de que se inscribieran 108 pacientes) cuando se demostró la superioridad de blinatumomab de acuerdo a una regla de suspensión preespecificada.
- La tasa de SSC a 24 meses fue del 66,2 % en los pacientes que recibieron blinatumomab, en comparación con el 27,1 % en los pacientes que recibieron el bloque de quimioterapia intensiva (cociente de riesgos instantáneos [CRI], 0,33; P< 0,001).
- La incidencia acumulada de recaída a 24 meses fue significativamente inferior en los pacientes que recibieron blinatumomab que en los pacientes que recibieron quimioterapia intensiva (24,9 vs. 70,8 %; CRI, 0,24).
- Entre los pacientes con ERM alta (≥10-4) en el momento que comenzó la aleatorización al tratamiento, un mayor número de pacientes presentó ERM negativa con blinatumomab que con el tratamiento de quimioterapia intensiva (93 vs. 24 %).
- En un ensayo clínico de fase III del COG se compararon 2 cursos de blinatumomab con 2 bloques de quimioterapia intensiva en pacientes con LLA-B en primera recaída. La aleatorización al tratamiento se hizo después de un único bloque de quimioterapia de reinducción intensiva. Los pacientes aptos para participar fueron aquellos con recaída temprana o aquellos con recaída tardía, pero con índices de ERM superiores al 0,1 % después de la quimioterapia de reinducción. La inscripción en el estudio terminó antes de tiempo debido a la pérdida de indeterminación clínica, a favor del blinatumomab. Esto fue determinado por el comité de vigilancia de datos e inocuidad del estudio. Los hallazgos principales de este ensayo son los siguientes:
- La tasa de SSC a 2 años fue del 54,4 % en los pacientes que recibieron blinatumomab versus el 39,0 % en los pacientes que recibieron quimioterapia (CRI de progresión de la enfermedad o mortalidad, 0,70; IC 95 %, 0,47–1,03).
- La tasa de SG fue del 71,3 % en los pacientes que recibieron blinatumomab versus el 58,4 % en los pacientes que recibieron quimioterapia (CRI de mortalidad, 0,62; IC 95 %, 0,39–0,98).
- Las tasas de ERN negativa fueron superiores en los pacientes después de 1 o 2 cursos de blinatumomab (75 y 66 %, respectivamente) que después de 1 o 2 cursos de quimioterapia intensiva (32 y 32 %, respectivamente).
- Las tasas de eventos adversos debidos a efectos tóxicos, como sepsis, mucositis o neutropenia febril, fueron mucho más bajas en los pacientes que recibieron blinatumomab que en los pacientes que recibieron quimioterapia intensiva.
Leucemia linfoblástica aguda de células B en recaída tardía
En estudios previos se observó que, en los pacientes de LLA-B con una recaída medular tardía, se obtenían tasas de supervivencia de alrededor del 50 % cuando se usaba un abordaje quimioterapéutico primario después de lograr la segunda RC, y no quedaba claro si el trasplante alogénico se relacionaba con una tasa de curación superior.; [Nivel de evidencia C1] A partir de datos posteriores, se demostró que la presencia de ERM al final de la reinducción permite identificar a los pacientes que tienen riesgo alto de recaída posterior, si se tratan con quimioterapia sola (sin TCMH) en el momento de la segunda RC. En varios estudios se observó que los pacientes con recaída medular tardía que tienen una ERM alta al final de la reinducción exhiben mejores desenlaces si reciben un TCMH alogénico en el momento de la segunda RC después de lograr un estado de ERM bajo o indetectable.
Evidencia (estratificación del riesgo de recaída tardía en la LLA-B a partir de la ERM):
- En los estudios del BFM, los pacientes se consideran como de riesgo intermedio si presentan una recaída medular aislada tardía o una recaída combinada medular y extramedular temprana o tardía. En el estudio ALL-REZ BFM P95/96 de este grupo, la ERM al final de la reinducción (evaluada mediante prueba de reacción en cadena de la polimerasa) fue un factor pronóstico significativo de los desenlaces en niños con riesgo intermedio de recaída de LLA-B tratados con quimioterapia sola en el momento de la segunda RC (sin TCMH).
- Los pacientes con ERM baja (<0,1 %) tuvieron una SSC a 10 años del 73 %, mientras que aquellos con ERM alta (≥0,1 %) tuvieron una SSC a 10 años del 10 %. En el análisis multivariante, la ERM al final de la reinducción fue el factor pronóstico independiente más sólido.
- En un estudio posterior del BFM (ALL-REZ BFM 2002 [NCT00114348]), los pacientes con riesgo intermedio de recaída se asignaron a un grupo que recibió un TCMH alogénico si presentaban ERM alta al final del primer mes de tratamiento. Los pacientes que tuvieron una ERM baja al final de la reinducción se trataron con quimioterapia sola (sin TCMH).
- La tasa de SSC fue del 64 % en los pacientes con ERM alta al final de la reinducción tratados con TCMH alogénico en la segunda RC; este resultado fue significativamente superior al del ensayo anterior P95/96, durante el cual los pacientes recibieron quimioterapia sin TCMH. La mejora en la SSC se debió sobre todo a un riesgo significativamente más bajo de recaída en la cohorte sometida a TCMH en el momento de la segunda RC (incidencia acumulada de recaída del 27 % en el ensayo de 2002, comparada con el 59 % en el ensayo P95/96).
- Los pacientes con recaídas medulares tardías y ERM baja al final de la reinducción que se trataron con quimioterapia sola tuvieron una SSC a 5 años del 76 %, lo que confirma los resultados observados antes en el ensayo P95/96. Sin embargo, la estrategia de quimioterapia sola produjo un desenlace significativamente más precario en los pacientes con recaídas combinadas tempranas (medulares y extramedulares) y ERM baja al final de la reinducción. La tasa de SSC a 5 años en estos pacientes fue solo del 37 %. A partir de estos resultados, los pacientes con recaídas combinadas tempranas ahora se clasifican como pacientes de riesgo alto en los estudios del grupo BMF.
- En los pacientes con recaídas medulares tardías y una ERM alta al final de la reinducción (definida como ≥0,1 %), el índice de la ERM fue importante para el pronóstico. La tasa de SSC a 10 años fue significativamente inferior en aquellos que tuvieron índices de ERM ≥1 %, en comparación con los que tuvieron índices de ERM ≥0,1 % a <1 % (56 vs. 74 %, respectivamente; P = 0,02). Por el contrario, para los pacientes con ERM baja al final de la reinducción (<0,1 %), no hubo ninguna diferencia significativa entre los que tuvieron una ERM muy baja (<0,01 %) y los que tuvieron índices de ERM de entre 0,01 % y <0,1 %.
- En el ensayo ALLR3 del Reino Unido, los pacientes que recayeron después de más de 6 meses de terminar el tratamiento inicial se asignaron a recibir un TCMH si la ERM al final de la reinducción era ≥0,01 % o a quimioterapia si la ERM era <0,01 %.
- De los 228 pacientes tratados, 220 pacientes lograron RC; 127 pacientes se asignaron a un TCMH (ERM alta) y 93 pacientes se asignaron a quimioterapia (ERM baja o no evaluable). Las tasas de SSC a 5 años fueron del 72 % en los pacientes con ERM baja versus el 56 % en los pacientes con ERM alta. Las tasas de SG a 5 años fueron del 87 % en los pacientes con ERM baja y del 64 % en los pacientes con ERM alta al final de la inducción.
- Sobre la base de estos datos, los investigadores del Reino Unido recomiendan un TCMH para los pacientes con LLA-B en recaída tardía que tienen ERM ≥0,01 % después del tratamiento de reinducción y quimioterapia sola para los que tienen una ERM al final de la reinducción <0,01 %.
- En el ensayo AALL1331 (NCT02883049) del COG, los pacientes con LLA de riesgo bajo en recaída se asignaron al azar a recibir quimioterapia estándar o quimioterapia estándar con 3 bloques de blinatumomab que remplazaron un bloque de quimioterapia intensiva. El riesgo bajo se definió como grado de ERM al final de la inducción <0,1 % en pacientes con recaída en la médula ósea que se presenta más de 36 meses después del diagnóstico o recaída extramedular que se presenta más de 18 meses después del diagnóstico.
- Para los 255 pacientes del protocolo, las tasas de SSE a 4 años fueron del 61,2 % en los pacientes que recibieron blinatumomab y del 49,5 % en los pacientes que recibieron quimioterapia estándar (P = 0,0849). Las tasas de SG a 4 años fueron del 90,4 % en los pacientes que recibieron blinatumomab, en comparación con el 79,6 % en los pacientes que recibieron quimioterapia estándar (P = 0,11).
- En los pacientes con recaída en la médula ósea con compromiso extramedular o sin este, las tasas de SSE a 4 años fueron del 72,7 % en los pacientes que recibieron blinatumomab y del 53,7 % en aquellos que recibieron quimioterapia estándar (P = 0,015). Las tasas de SG a 4 años fueron del 97,1 % en los pacientes que recibieron blinatumomab, en comparación con el 84,8 % en el grupo de quimioterapia sola (P = 0,02).
- En los pacientes con recaída extramedular aislada, los desenlaces fueron precarios, con independencia del grupo de tratamiento. Los desenlaces de estos pacientes fueron peores a los observados en pacientes similares en ensayos anteriores. Las tasas de SSE a 4 años fueron del 36,6 % en los pacientes que recibieron blinatumomab y del 38,8 % en los que recibieron quimioterapia sola (P = 0,62). Las tasas de SG a 4 años fueron del 76,5 % en el grupo de blinatumomab, en comparación con el 68,8 % en el grupo de quimioterapia sola (P = 0,53).
- En el ensayo del COG AALL0433 (NCT00381680) se incluyeron pacientes con LLA-B en recaída de riesgo intermedio (definida como recaída tardía con afectación medular que ocurre >36 meses desde el diagnóstico inicial, o recaída extramedular aislada muy temprana que ocurre <18 meses desde el diagnóstico inicial). Los pacientes recibieron 3 bloques de inducción, seguidos de una quimioterapia intensiva de base (con radioterapia en los pacientes con afectación testicular o del SNC) o TCMH alogénico (si había donantes fraternos compatibles). En el ensayo también se incluyó una comparación aleatorizada de dosificación estándar versus intensificada de vincristina. De los 271 pacientes aptos, 257 (95 %) alcanzaron una segunda RC. De esos pacientes, 74 (29 % de aquellos en segunda RC) recibieron TCMH; entre ellos, 47 tenían donantes fraternos compatibles y 27 tenían otros donantes.
- Las tasas de SSC a 3 años y de SG en los 271 pacientes aptos fueron del 63,6 % y del 72,3 %, respectivamente.
- En los pacientes con recaída medular tardía (n = 242), las tasas de SSC a 3 años y de SG fueron del 66,3 % y del 74,8 %, respectivamente.
- En los pacientes con recaída medular tardía, la ERM al final del primer mes de tratamiento (ERM al final de la reinducción) fue un factor pronóstico importante. En los pacientes con ERM al final de la reinducción inferior al 0,1 %, la tasa de SSC a 3 años fue del 84,9 % versus el 53,7 % en aquellos con ERM superior al 0,1 %.
- Al hacer el ajuste por mediana de tiempo para el TCMH, los pacientes que recibieron TCMH presentaron una tasa de SSE a 3 años superior (P = 0,03), pero no una tasa de SG superior (P = .46), en comparación con los pacientes que recibieron quimioterapia sola.
- La asignación al azar a vincristina se interrumpió de forma prematura debido a un exceso de toxicidad, sobre todo en pacientes de edad más avanzada.
Leucemia linfoblástica aguda positiva para BCR::ABL1 (positiva para el cromosoma Filadelfia [Ph+])
Hay poca información sobre el tratamiento de pacientes con LLA BCR::ABL1 en la era de los inhibidores de tirosina–cinasas (ITC).
En un estudio multicéntrico francés se notificaron 27 niños con LLA BCR::ABL1 en recaída (24 evidentes, 3 moleculares) que inicialmente habían sido tratados con un régimen que incluyó imatinib.[Nivel de evidencia C1]
- El 96 % de los pacientes que se trataron con un régimen de inducción intensivo o uno no intensivo alcanzaron una segunda RC.
- Alrededor de la mitad de los pacientes recibieron terapia de consolidación y el 78 % se sometieron a TCMH.
- En el momento de la recaída, el ITC se modificó en 23 de los 26 (88 %) pacientes, y 15 de ellos (58 %) recibieron dasatinib. La tasa de SSC a 4 años fue del 60,9%, y la tasa de SG fue del 76,1%.
Leucemia linfoblástica aguda de células T
En los pacientes con LLA-T que alcanzaron la remisión después de una recaída en la médula ósea, los desenlaces con quimioterapia posreinducción sola han sido por lo general precarios, y estos pacientes a menudo se tratan con TCMH alogénico en el momento de la segunda RC, sin tener en cuenta el tiempo hasta la recaída. En los pacientes con LLA-T en segunda remisión la tasa de SG a los 3 años de un trasplante alogénico fue del 48 % y la tasa de SSE fue del 46 %.[Nivel de evidencia C1]
Opciones de tratamiento para la segunda recaída en la médula ósea y recaídas posteriores
Aunque no hay estudios de comparación directa entre la quimioterapia y el TCMH para los pacientes que alcanzan una RC por tercera vez o posterior, el trasplante por lo general se considera un abordaje razonable para quienes alcanzan la remisión porque la curación con quimioterapia sola es infrecuente. La supervivencia a largo plazo para los pacientes de LLA después de la segunda recaída es especialmente precaria, del orden de menos del 10 % al 20 %. Una de las principales razones de este fracaso es la imposibilidad de lograr una tercera remisión. Numerosos intentos de abordajes con combinaciones nuevas se han traducido en que solo cerca del 40 % de los niños logra una remisión durante la segunda recaída. Sin embargo, en 2 estudios se añadió el bortezomib a los fármacos de reinducción estándar para pacientes con múltiples recaídas resistentes al tratamiento y se obtuvieron tasas de RC del 70 % al 80 %.[Nivel de evidencia C1]; [Nivel de evidencia C3] La clofarabina en monoterapia también se ha usado en pacientes con múltiples recaídas y leucemia resistente al tratamiento. En un análisis de 12 estudios clínicos de clofarabina en pacientes pediátricos con LLA en recaída o resistente al tratamiento, la tasa de remisión general (RC y RC con recuperación incompleta de plaquetas o recuperación incompleta de neutrófilos y plaquetas) fue del 28 %.
En un ensayo de fase I se probó la combinación de venetoclax (inhibidor de BCL2) y navitoclax (inhibidor de BCL2 y BCL-XL) administrados junto con quimioterapia estándar (vincristina, dexametasona, con pegaspargasa o sin esta) en pacientes adultos y pediátricos con LLA-B o LLA-T en múltiples recaídas o resistente al tratamiento. En general, la combinación se toleró bien (el principal efecto tóxico fue la mielodepresión prolongada) y se logró la RC en el 60 % de los pacientes; el 57 % de ellos tuvo ERM indetectable.
En los múltiples pacientes en recaída que logran una RC, se ha observado que el TCMH cura de un 20 % a un 35 % de los casos y que los fracasos obedecen a tasas altas de recaída y mortalidad relacionada con el trasplante.[Nivel de evidencia C1]
Debido a los desenlaces precarios en los pacientes de LLA-B con recaídas múltiples que reciben quimioterapia seguida de TCMH, la terapia de células T con CAR se ha usado como tratamiento estándar en esta población y se han obtenido tasas altas de remisión, así como una supervivencia superior (a pesar de que hacen falta ensayos comparativos directos). Para obtener más información, consultar la sección Terapia de células T con receptor de antígeno quimérico.
Los fármacos de inmunoterapia como blinatumomab e inotuzumab se han usado en esta población y han mejorado las tasas de remisión; a menudo se ha logrado la curación cuando a continuación se hace un TCMH. Todavía no se han llevado a cabo estudios comparativos de abordajes de inmunoterapia y terapia celular en esta población, de manera que no hay datos para determinar los abordajes óptimos para el tratamiento inicial ni la secuencia de tratamientos.
Trasplante de células madre hematopoyéticas en la primera recaída medular y en recaídas posteriores
Componentes del proceso de trasplante
En 2012, se publicó una revisión de expertos sobre las indicaciones para el TCMH. Los componentes del proceso de trasplante que han mostrado ser importantes para mejorar o predecir el desenlace del TCMH en los niños con LLA, son los siguientes:
- Regímenes preparatorios para el trasplante con irradiación corporal total (ICT).
- Detección de ERM justo antes del trasplante.
- Detección de ERM después del trasplante
- Tipo de donante y compatibilidad de HLA.
- Función de la enfermedad de injerto contra huésped o injerto contra leucemia en la LLA y la modulación inmunitaria después del trasplante para evitar una recaída.
En un análisis del CIBMTR se evaluaron las variables previas al trasplante para crear un modelo con el fin de predecir la SSL posterior al trasplante en pacientes pediátricos (menores de 18 años). Todos los pacientes recibían un trasplante por primera vez y tenían acondicionamiento mielosupresor; se incluyeron todas las fuentes de células madre. En los pacientes con LLA, los factores predictivos relacionados con una SSL más baja fueron la edad menor de 2 años, una segunda RC o superior, la ERM positiva (solo en la segunda RC, no en la primera RC) y la presencia de enfermedad detectable mediante estudios morfológicos en el momento del trasplante. Se creó una escala para estratificar a los pacientes de acuerdo a los factores de riesgo con el fin de predecir la supervivencia. La tasa de SSL a 5 años fue del 68 % en el grupo de riesgo bajo, del 51 % en el grupo de riesgo intermedio y del 33 % en el grupo de riesgo alto.
Regímenes preparatorios para el trasplante con irradiación corporal total
La irradiación corporal total (ICT) es un componente importante del régimen de acondicionamiento para los pacientes que se someten a un TCMH alogénico. En varios estudios se notificó que la ICT se relaciona con desenlaces superiores en los pacientes con LLA, en comparación con los regímenes preparatorios de quimioterapia sola.
Evidencia (ICT como parte del régimen preparatorio en la LLA):
- En 2 estudios de registros y en un ensayo aleatorizado pequeño se observó que los regímenes de acondicionamiento para el trasplante que incluyeron la ITC produjeron tasas de curación más altas que los regímenes preparatorios de quimioterapia sola.
- En un estudio internacional (Estados Unidos, Europa y Australia) en el que se combinaron conjuntos de datos de ensayos prospectivos y datos de centros individuales se observó que el uso de regímenes sin ICT fue un factor de riesgo independiente de un desenlace precario.
- En un ensayo de fase III internacional realizado entre 2013 y 2018 se incluyeron 417 pacientes pediátricos de 4 a 21 años con LLA. Los pacientes se asignaron al azar para recibir ICT (12 Gy) con etopósido o quimioterapia sola (fludarabina, tiotepa y busulfano o treosulfano) como régimen preparatorio. Todos los pacientes estaban en RC morfológica en el momento del trasplante y lo recibieron de un donante fraterno compatible 9/10 o 10/10 o de un donante no emparentado compatible. La idea original del estudio era incluir 1000 pacientes, pero se interrumpió la inscripción de forma prematura cuando se cumplió una regla de suspensión temprana por inutilidad, lo que indica que el régimen preparatorio de quimioterapia sola se relacionó con un desenlace más precario.
- La tasa de SG a 2 años fue del 91 % en los pacientes que recibieron un régimen que incluyó ICT, en comparación con el 75 % en los pacientes que recibieron un régimen preparatorio de quimioterapia sola (P < 0,001).
- La tasa de SSC a 2 años también fue superior en los pacientes que recibieron ICT (86 vs. 58 %, P< 0,001), sobre todo debido a una incidencia acumulada de recaída más baja en la cohorte de ICT (incidencia acumulada de recaída a 2 años, 12 % en los pacientes con ICT vs. 33 % en los pacientes con quimioterapia sola).
- En un análisis de subgrupos, el régimen de ICT fue superior, con independencia del tipo de donante (fraterno compatible vs. no emparentado compatible), el inmunofenotipo (LLA-B vs. LLA-T) o de si el trasplante se hizo en la primera o en la segunda RC.
- No hubo diferencias en las tasas de EICH aguda o crónica ni en la frecuencia de efectos adversos no hematológicos de grados 3 o 4 entre los 2 grupos aleatorizados de tratamiento.
Según estos datos, para casi todos los niños, excepto los más pequeños (edad <3 o 4 años), la ICT sigue siendo el tratamiento estándar en la mayoría de las instituciones en América del Norte y Europa.
La ITC fraccionada (dosis total, 12–14 Gy) a menudo se combina con ciclofosfamida, etopósido, tiotepa o una combinación de estos fármacos. Los hallazgos de los estudios de estas combinaciones produjeron, en general, tasas similares de supervivencia, aunque en un estudio se indicó que, si se usa la ciclofosfamida sin otros fármacos quimioterapéuticos, quizás se necesite una dosis de ITC más alta. Muchos regímenes estándar incluyen ciclofosfamida con dosis de ITC entre 13,2 y 14 Gy. Por otro lado, cuando se usaron ciclofosfamida y etopósido con ICT, las dosis superiores a 12 Gy produjeron una supervivencia más precaria debido a una toxicidad excesiva.
En un análisis secundario del ensayo ASCT0431 (NCT00382109) de TCMH se demostró que todos los pacientes tratados con ICT que incluyó modulación de la dosis en los campos pulmonares a menos de 8 Gy tuvieron una ventaja de supervivencia en el análisis multivariante (cociente de riesgos instantáneos [CRI], 1,85; P = 0,04). La mortalidad relacionada con el trasplante tendió a ser más alta en los pacientes que recibieron dosis de 8 Gy o superiores, pero no alcanzó significación estadística (CRI, 1,78; P = 0,21). Debido a que las dosis inferiores no se relacionaron con un aumento de las recaídas y mejoraron la supervivencia, se incluyó la modulación de la dosis a menos 8 Gy en campos pulmonares en el ensayo AALL1331 (NCT02883049) del COG. Se necesita contar con los resultados del estudio AALL1331 y otros estudios que analicen con mayor precisión la modulación de la dosis pulmonar de ICT para aclarar y explicar esta observación.
Detección de la enfermedad residual mínima justo antes del trasplante
Desde hace tiempo se sabe que el estado de la remisión en el momento del trasplante es un factor importante para predecir el desenlace; los pacientes que no se encuentran en un periodo de RC en el momento del TCMH tienen tasas de supervivencia muy precarias. En varios estudios también se demostró que un índice de ERM alto en el momento del trasplante es un factor de riesgo importante en niños con LLA en RC que se someten a un TCMH alogénico.[Nivel de evidencia C1]; Se ha informado que las tasas de supervivencia en los pacientes positivos para la ERM antes del trasplante oscilan entre el 20 % y el 47 %, en comparación con el 60 % al 88 % en los pacientes negativos para la ERM.
Cuando los pacientes reciben 2 a 3 ciclos de quimioterapia en un intento por lograr una remisión negativa para la ERM, el beneficio de un tratamiento más intensivo para lograr la desaparición de la ERM se debe sopesar con la posibilidad de producir toxicidad grave. Además, no hay datos claros que indiquen que un resultado positivo para la ERM en un paciente que ha recibido múltiples ciclos de tratamiento sea un marcador biológico de la enfermedad que permita predecir un desenlace precario inmodificable, o si una intervención adicional que lleve a estos pacientes a una remisión negativa para la ERM superaría este factor de riesgo y mejoraría la supervivencia.
- En un informe, 13 pacientes de LLA con ERM alta en el momento del trasplante planificado recibieron un ciclo adicional de quimioterapia en un intento de disminuir la ERM antes de proceder al TCMH. Luego del TCMH, 10 de 13 pacientes (77 %) permanecieron en RC, y no se presentaron recaídas en los 8 pacientes que alcanzaron una ERM baja luego del ciclo adicional de quimioterapia. En comparación, solo 6 de 21 pacientes con ERM alta (29 %) que se sometieron directamente al TCMH sin recibir quimioterapia adicional antes del trasplante, permanecieron en RC.
Detección de la enfermedad residual mínima después del trasplante
La presencia de ERM detectable luego de un TCMH acarrea un riesgo alto de recaída subsiguiente. En los pacientes con ERM detectable antes del TCMH, la detección de cualquier grado de ERM después del TCMH pone al paciente en riesgo muy alto de fracaso (>90 %). La exactitud de la ERM para predecir la recaída aumenta a medida que pasa tiempo desde el TCMH, y el riesgo de recaída también es más alto para los pacientes que exhiben índices más altos de ERM en cualquier momento. En un estudio se observó una sensibilidad más alta para predecir la recaída de los análisis de secuenciación de última generación en lugar de la citometría de flujo, en especial, poco tiempo después del TCMH.
Tipo de donante y compatibilidad de HLA
Las tasas de supervivencia después de un trasplante de un donante no emparentado compatible y de un trasplante de sangre de cordón umbilical han mejorado bastante en la última década y ofrecen un desenlace similar al obtenido con trasplantes de un donante fraterno compatible.; [Nivel de evidencia B4]; [Nivel de evidencia C1]; [Nivel de evidencia C2] Las tasas de EICH clínicamente extensa y de la mortalidad relacionada con el tratamiento siguen siendo más altas después de un trasplante de un donante no emparentado, en comparación con trasplantes de un donante fraterno compatible. Sin embargo, hay evidencia que indica que el trasplante de donante no emparentado compatible quizás produzca tasas de recaída más bajas. Los análisis del National Marrow Donor Program y del CIBMTR demostraron que las tasas de EICH, mortalidad relacionada con el tratamiento y SG han mejorado a lo largo del tiempo.; [Nivel de evidencia C1]
En otro estudio del CIBMTR, se indicó que el desenlace después de trasplantes de sangre del cordón umbilical con incompatibilidad de 1 o 2 antígenos quizás sea equivalente al de un trasplante de un donante emparentado compatible o un donante no emparentado compatible. En ciertos casos en los que no hay un donante compatible, o cuando se considera que se necesita un trasplante inmediato, es posible considerar un trasplante haploidéntico con concentraciones altas de células madre. Con los abordajes mejorados de TCMH haploidénticos en los que se mide la reducción de receptor de célula T α-β (TCR)/CD19 o se usa ciclofosfamida después del trasplante, se observan tasas de supervivencia similares a las de estudios en los que se usan otras fuentes de células madre. En un ensayo multicéntrico grande de Italia se observaron desenlaces similares cuando se usaron donantes haploidénticos con reducción de TCR α-β/CD19, en comparación con donantes no emparentados compatibles, pero con tasas más bajas de EICH. En un segundo ensayo multicéntrico, en el que se usaron donantes haploidénticos favorables para el receptor similar a la inmunoglobulina de células citolíticas naturales (KIR) TCR α-β/CD19, se observaron resultados de supervivencia comparables a todas las demás fuentes de células madre, con tasas más bajas de EICH y de mortalidad relacionada con el trasplante.
Función de la enfermedad de injerto contra huésped o injerto contra leucemia en la leucemia linfoblástica aguda y la modulación inmunitaria después del trasplante para evitar una recaída
La mayoría de los estudios que abordan este tema en pacientes pediátricos y adultos jóvenes indican un efecto de la enfermedad de injerto contra huésped (EICH) aguda y crónica en la reducción de las recaídas.
- En un ensayo del COG sobre trasplantes en niños con LLA, los grados l a III de EICH aguda se relacionaron con un riesgo más bajo de recaída (CRI, 0,4; P = 0,04) y mejor SSC (análisis multivariante, CRI, 0,5; P = 0,02). El aumento acentuado de la mortalidad relacionada con el trasplante (CRI, 6,4; P = 0,003) contrarrestó cualquier efecto de la EICH aguda de grado IV en la disminución del riesgo de recaída, mientras que la EICH aguda de grados l a III no tuvo ningún efecto detectable estadísticamente significativo en la mortalidad relacionada con el trasplante (CRI, 0,6; P = 0,42).
- En un modelo multivariante, la ERM antes del trasplante y la EICH aguda fueron factores de predicción independientes de recaída; el riesgo más bajo de recaída se observó en pacientes con una ERM pretrasplante baja y una EICH aguda de grado l a III. En los pacientes que no presentaron EICH aguda hasta el día 55 después del TCMH, casi todas las recaídas se presentaron entre los días 100 y 400 después del TCMH.
Aprovechando este efecto de ICL, se han estudiado varios abordajes para prevenir la recaída después del trasplante, como la interrupción de la depresión inmunitaria o la infusión de linfocitos de donantes y las inmunoterapias dirigidas, entre ellas, el uso de anticuerpos monoclonales y células citolíticas naturales. En algunos ensayos de Europa y los Estados Unidos, se observó que los pacientes en riesgo alto de recaída según un aumento de quimerismo del receptor (es decir, el aumento del porcentaje de marcadores de DNA del receptor) logran de manera exitosa una interrupción de la depresión inmunitaria sin toxicidad excesiva.
- En un estudio se observó que, de 46 pacientes con aumento de quimerismo del receptor, los 31 pacientes que interrumpieron la depresión inmunitaria y recibieron una infusión de linfocitos de un donante o recibieron ambos tratamientos tuvieron una SSC a 3 años del 37 % versus el 0 % en el grupo sin intervención (P< 0,001).
- En otros estudios, se observaron mejores tasas de supervivencia de las esperadas en los pacientes con ERM antes del TCMH cuando se redujeron los inmunodepresores en casos de ERM detectada luego del TCMH.
- En un estudio internacional extenso se mostró una disminución marcada en la recaída de pacientes con ERM después del TCMH que presentaban una EICH aguda (CRI, 0,29; P< 0,001), lo cual mejoró la SSC (CRI, 2,9; P = 0,01). La EICH aguda disminuyó en forma significativa la recaída y mejoró la SSC en los pacientes positivos para la ERM y en aquellos negativos para la ERM. La EICH crónica también se relacionó con menos recaídas en pacientes positivos para la ERM y en pacientes negativos para la ERM.
Medicamentos intratecales después de un trasplante de células madre hematopoyéticas para prevenir la recaída
El uso de la quimioprofilaxis con quimioterapia intratecal después del TCMH es polémico.
Recaída después de un trasplante de células madre hematopoyéticas alogénico en la leucemia linfoblástica aguda en recaída
En los pacientes con LLA-B que recaen después de un trasplante de células madre hematopoyéticas (TCMH) alogénico, en quienes se puede retirar de manera gradual el tratamiento inmunodepresor y que no tienen EICH, se observó que el uso de tisagenlecleucel o de otros abordajes de células T con CAR 4-1BB produjo tasas de SSC que superaron el 50 % a los 12 meses. En los pacientes con LLA-T que recayeron o en los pacientes con LLA-B que no son aptos para recibir la terapia de células T con CAR, quizás sea viable un segundo TCMH alogénico ablativo. Sin embargo, muchos pacientes no se podrán someter a un segundo procedimiento de TCMH debido a la incapacidad de alcanzar la remisión, muerte prematura por toxicidad o toxicidad orgánica grave relacionada con la quimioterapia de rescate. Entre el grupo de pacientes seleccionados que son aptos para recibir un segundo TCMH alogénico ablativo, de un 10 % a un 30 % alcanzará una SSC a largo plazo.; [Nivel de evidencia C1] El pronóstico es más favorable en los pacientes con una remisión más prolongada después del primer TCMH y en aquellos con RC en el momento del segundo TCMH. Además, en un estudio se observó un aumento de la supervivencia después del segundo TCMH si se presentaba EICH aguda, en especial, si esta no se había presentado después del primer trasplante.
Los abordajes de intensidad reducida también curan un porcentaje de pacientes cuando se usan con un segundo trasplante alogénico, pero solo si los pacientes logran una RC confirmada por citometría de flujo.[Nivel de evidencia B4] La infusión de leucocitos de un donante tiene pocos beneficios para los pacientes con LLA que recaen después de un TCMH alogénico.; [Nivel de evidencia C1]
Se desconoce si es necesario un segundo trasplante alogénico para tratar la recaída aislada en el SNC o en los testículos después de un TCMH. En una serie pequeña se observó supervivencia en algunos pacientes que recibieron quimioterapia sola o quimioterapia seguida de un segundo trasplante.[Nivel de evidencia C1]
Abordajes inmunoterapéuticos para la leucemia linfoblástica aguda en recaída o resistente al tratamiento
Los abordajes inmunoterapéuticos para la LLA en recaída o resistente al tratamiento incluyen los anticuerpos monoclonales y las células T con CAR.
Terapia con anticuerpos monoclonales
Se han estudiado los siguientes 2 fármacos inmunoterapéuticos para la LLA-B en recaída o resistente al tratamiento:
- Blinatumomab. El blinatumomab es un anticuerpo monoclonal biespecífico con un sitio de unión a CD3 (células T) y otro sitio de unión a CD19 (presente en la mayoría de las células de la LLA-B). Por lo tanto, el blinatumomab promueve la unión de las células T citotóxicas del paciente a los linfoblastos B, lo que conlleva la destrucción del tumor.
En un ensayo de fase I/II de niños menores de 18 años con LLA-B en recaída o resistente al tratamiento, 27 de 70 pacientes (39 %) tratados con blinatumomab en monoterapia en la dosis recomendada para la fase II lograron una RC; el 52 % de los pacientes que lograron una RC no presentaron ERM.
En un análisis conjunto de 5 ensayos que incluyeron 166 pacientes pediátricos y 517 adultos, aquellos con menos del 50 % de blastocitos en la médula ósea al inicio del tratamiento tuvieron mejores respuestas al blinatumomab. Entre los pacientes pediátricos, las tasas de RC (incluye la RC con recuperación hematológica parcial [RCh] y la RC con recuperación hematológica incompleta [RCi]) fueron del 65,3 % en los pacientes con un porcentaje inicial de blastocitos en médula ósea menor al 50 %, en comparación con el 38,3 % en los pacientes con un porcentaje inicial de blastocitos en médula ósea del 50 % o más. De manera similar, las respuestas de ERM fueron más frecuentes en pacientes con un porcentaje inicial de blastocitos en médula ósea menor al 50 %, que en aquellos que tenían un porcentaje inicial de blastocitos en médula ósea del 50 % o más (51,4 vs. 25,5 %). No hubo diferencias significativas en estos criterios de valoración al comparar los pacientes que tenían entre un 5 % y menos de un 25 % de blastocitos al inicio del tratamiento con blinatumomab, y aquellos con entre un 25 % y menos de un 50 %.
- Inotuzumab. El inotuzumab es un anticuerpo monoclonal anti-CD22 que se conjuga con la caliqueamicina.
En ensayos de pacientes adultos con LLA de células B en recaída o resistente al tratamiento se logró una RC en casi el 80 % de los pacientes.
Se han realizado 2 ensayos de fase II de inotuzumab en monoterapia (en ambos ensayos se usó una dosis total de 1,8 mg/m2 en el primer curso y de 1,5 mg/m2 en los cursos posteriores) para el tratamiento de pacientes pediátricos con LLA en recaída o resistente al tratamiento.
- En el ensayo de fase II del COG (N = 48), la tasa de respuesta general (RC + RCi) fue del 58,3 % después del primer curso y del 62,5 % después de 2 cursos. Entre los pacientes que alcanzaron una respuesta completa o una respuesta completa con recuperación incompleta del recuento, el 70 % presentó ERM negativa (<0,01 %). En general, el inotuzumab se toleró bien, y no se presentó hepatotoxicidad importante ni síndrome de obstrucción sinusoidal (SOS) durante el tratamiento. Sin embargo, de 21 pacientes que posteriormente se sometieron a TCMH, 6 (28.6%) presentaron SOS.
- En un ensayo de fase II del grupo Innovative Therapies for Children With Cancer (N = 28), la tasa de respuesta general fue del 81,5 % (todas las respuestas ocurrieron después del primer ciclo). De los pacientes que alcanzaron una respuesta, el 81,8 % alcanzó una ERM negativa (<0,01 %), el 59,1 % después del primer curso y el resto de los pacientes después del segundo curso. De los pacientes, 7 (28 %) presentaron SOS, 6 de ellos después de un TCMH posterior y 1 durante el tratamiento con inotuzumab. No hubo ninguna correlación entre el grado de expresión de CD22 (media del índice de fluorescencia o porcentaje de células positivas para CD22) y la respuesta. En un análisis posterior, en el que se combinaron pacientes de los ensayos de fase I y fase II, los autores encontraron que el intervalo menor entre la última dosis de inotuzumab y el TCMH se asociaba con un mayor riesgo de presentar SOS.
Las recomendaciones de un grupo de expertos para la prevención del SOS relacionado con el TCMH después del uso de inotuzumab incluyen que el tratamiento se limite a 2 cursos de inotuzumab, se eviten los regímenes de TCMH con 2 alquilantes, se eviten los fármacos hepatotóxicos y se considere el uso de fármacos profilácticos para el SOS.
- Rituximab u ofatumumab. Estos anticuerpos monoclonales se dirigen a CD20, que se expresa de forma débil en los niños más pequeños con LLA, pero es más común en adolescentes y adultos jóvenes. En varios estudios se ha indicado algún beneficio cuando se añade uno de estos fármacos a la quimioterapia de reinducción en adultos, pero hay pocos datos publicados sobre niños.
Terapia de células T con receptor de antígeno quimérico
La terapia de células T con receptor de antígeno quimérico (CAR) es una estrategia terapéutica para los pacientes con LLA-B infantil y enfermedad resistente al tratamiento, o aquellos que se encuentran en una segunda recaída o recaídas posteriores. Este tratamiento implica la producción de células T con un CAR que logra cambiar la especificidad y el funcionamiento de las células T. El antígeno CD19 es una de las dianas moleculares que más se utiliza para las células T modificadas con un CAR; casi todas las células B normales expresan este antígeno, así como la mayoría de las células B malignas.
Toxicidad de la terapia de células T con receptor de antígeno quimérico
El tratamiento de células T con receptor de antígeno quimérico (CAR) se relacionó con un síndrome de liberación de citocinas que es potencialmente mortal. El síndrome de liberación de citocinas se manifiesta con fiebre, cefalea, mialgias, hipotensión, extravasación capilar, hipoxia y disfunción renal. El síndrome de liberación de citocinas grave se ha tratado de manera eficaz con tocilizumab, un anticuerpo anti-receptor de la interleucina-6 (IL-6R). La persistencia a largo plazo de las células T con CAR a veces produce aplasia de las células B, lo que conduce a la necesidad de reemplazar las inmunoglobulinas.
La terapia de células T con CAR también produce neurotoxicidad, afasia, alteración del estado mental y convulsiones; los síntomas se suelen resolver de modo espontáneo. Los síntomas en el sistema nervioso central no mejoraron con las sustancias dirigidas a IL-6R ni con otros abordajes.
Otros efectos secundarios de la terapia de células T con CAR incluyen coagulopatía, cambios de laboratorio similares a la linfohistiocitosis hemofagocítica y disfunción cardiaca. Entre un 20 % y un 40 % de los pacientes necesitan recibir tratamiento, que en su mayoría es apoyo con vasopresores, en la unidad de cuidados intensivos y un 10 % a un 20 % de los pacientes requieren intubación o diálisis. En un estudio, los pacientes que presentaron efectos tóxicos similares a linfohistiocitosis después de la terapia de células T con CAR presentaron tasas más bajas de supervivencia sin recaída (SSR) y SG en comparación con quienes no presentaron dichos efectos tóxicos (tasas de SSR de 25,7 % vs. 57,6 %; tasas de SG, 4 % vs. 81 %).
Para obtener una descripción más detallada de los efectos tóxicos de la terapia de células T con CAR y los abordajes para mitigar dichos efectos, consultar Terapia de células T con CAR en pediatría.
Terapia de células T con receptor de antígeno quimérico dirigido al CD19
Se han llevado a cabo varios ensayos clínicos de células T con receptor de antígeno quimérico (CAR) dirigido al CD19 para la LLA en recaída o resistente al tratamiento y los resultados son prometedores. Los ensayos publicados han incorporado el uso de 2 tipos de moléculas coestimuladoras, 4-1BB y CD28. Los abordajes basados en CD28 han generado tasas altas de remisión, pero las células T con CAR en estos ensayos casi nunca persisten más de 1 a 2 meses, por lo que se necesita de un TCMH para la supervivencia a largo plazo. En muchos de los ensayos en los que se usó coestimulación de 4-1BB hubo persistencia de las células T con CAR durante periodos extensos y respuestas a largo plazo. Dado que los informes preliminares sobre la eficacia eran mucho mejores que las experiencias anteriores, los ensayos que derivaron en la aprobación regulatoria del tisagenlecleucel para la LLA con múltiples recaídas o resistencias a tratamientos se llevaron adelante sin aleatorización; su diseño estadístico se basó en experiencias anteriores. En los análisis retrospectivos emparejados con controles en los que se usaron cohortes de tratamiento estándar anteriores a la terapia de células T con CAR se observaron importantes mejoras en la SG y en la SSC con el uso de tisagenlecleucel. Sin embargo, hasta ahora, en ningún ensayo prospectivo se han comparado los abordajes de células T con CAR y otras modalidades de tratamiento.
Evidencia (terapia de células T con CAR dirigido a CD19):
- En ensayos clínicos piloto del Children’s Hospital of Philadelphia (CHOP) y el Hospital of the University of Pennsylvania, 30 niños y adultos (25 de ellos de 22 años o menos) con LLA positiva para CD19 con múltiples recaídas o resistente al tratamiento recibieron células T sometidas a transducción mediante un vector lentivírico de un CAR con coestimulación de 4-1BB dirigido a CD19.[Nivel de evidencia C2]
- Se obtuvo una RC en el 90 % de los pacientes, entre ellos, 15 de 18 pacientes (83 %) que habían recibido previamente un TCMH alogénico.
- La tasa de SSC a 6 meses fue del 67 % y la mayoría de los pacientes exhibió persistencia de las células T con CAR y aplasia de células B durante 6 meses.
- Los 30 pacientes presentaron algún grado de síndrome de liberación de citocinas. De los pacientes, 8 (27 %) tuvieron síntomas graves y necesitaron tratamiento con vasopresores y apoyo respiratorio. El síndrome de liberación de citocinas se trató de manera eficaz con tocilizumab.
- En un informe de un ensayo de fase I, 45 niños y adultos jóvenes con LLA-B positiva para CD19 en recaída o con resistencia al tratamiento recibieron células T con CAR modificadas con un vector lentivírico 4-1BB, y se observaron los resultados que se listan a continuación.
- Una tasa de remisión general del 89 % en todos los pacientes inscritos en el análisis por intensión de tratar.
- Mejora de la persistencia a largo plazo de las células T con CAR y aplasia de las células B en los siguientes pacientes: 1) aquellos que se sometieron a estrategias de disminución de linfocitos utilizando fludarabina y ciclofosfamida, y 2) aquellos que comenzaron el tratamiento con un porcentaje más alto de células con expresión de CD19 (blastocitos o células B normales).
- A partir de los resultados de un ensayo mundial de fase II del vector anti-CD19 4-1BB formulado por CHOP y la University of Pennsylvania, la Administración de Alimentos y Medicamentos de los Estados Unidos aprobó el tisagenlecleucel para niños con LLA-B en recaídas múltiples o con resistencia al tratamiento.
- De los 92 pacientes inscritos, 75 recibieron una infusión de células T con CAR elaborada de manera exitosa. De los pacientes que recibieron la infusión, el 81 % presentó 2 mediciones en las que se observó RC en el transcurso de los primeros 3 meses desde la infusión, y el 100 % de las remisiones fueron negativas para la ERM.
- La tasa de SSC de los pacientes que recibieron la infusión fue del 73 % a los 6 meses y del 50 % a los 12 meses. La tasa de SG de los pacientes que recibieron la infusión fue del 90 % a los 6 meses y del 70 % a los 12 meses.
- El CIBMTR hizo un análisis de datos de la vida real de 255 pacientes con LLA que duplicó en gran medida los resultados que se observaron en ensayos fundamentales para el registro, con una tasa inicial de RC del 85,5 %. La tasa de respuesta de 12 meses de duración fue del 69,9 %, la tasa de SSC fue del 52,4 %, y la tasa de SG fue del 77,2 %.
- No se incluyeron niños menores de 3 años en el ensayo para la aprobación del tisagenlecleucel. Un subgrupo de pacientes, los lactantes que reciben el diagnóstico antes del año de edad y que tienen una leucemia con reordenamiento de KMT2A, suelen presentar una enfermedad muy resistente a los abordajes de quimioterapia intensivos. En un estudio multicéntrico retrospectivo se notificaron los desenlaces para los niños menores de 3 años (cuyo diagnóstico original fue de LLA de alto riesgo en lactantes) con LLA-B en recaída o resistente al tratamiento que recibieron tisagenlecleucel comercial.
- Se seleccionaron 38 pacientes, de los cuales 35 (92 %) recibieron la infusión de tisagenlecleucel (2 pacientes no recibieron la infusión por defecto en la fabricación de las células T con CAR T y el otro paciente presentó progresión de la enfermedad).
- De los 38 pacientes, 29 (76 %) tenían reordenamientos de KMT2A y 25 (66 %) presentaron recidiva después de un TCMH previo.
- Al cabo de una mediana de seguimiento de 14 meses, la tasa de SG a 12 meses fue del 84 %, la tasa de SSP del 69 % y la tasa de SSP estricta fue del 41 %.
- La viabilidad, los desenlaces y la toxicidad fueron comparables con los resultados de niños mayores.
- En un estudio posterior de la Universidad de Pensilvania se notificaron los resultados actualizados a 3 años del ensayo ELIANA (NCT02435849) realizado a nivel mundial en el que se incluyeron 79 pacientes que se trataron con tisagenlecleucel.[Nivel de evidencia C2]
- La tasa de remisión general fue del 82 % a los 3 meses.
- Al cabo de una mediana de seguimiento de 38,8 meses, no se alcanzó la mediana de duración de la remisión de los 66 pacientes con una respuesta completa. La tasa de supervivencia sin recaída (SSR) fue del 58 % a los 24 meses y del 52 % a los 36 meses.
- De los 32 pacientes con RC que no recibieron tratamiento adicional, la tasa de SSR fue del 81 % a los 24 meses y del 76 % a los 36 meses.
- En un informe de la Pediatric Oncology Branch del NCI, se describió el uso de un producto diferente de células T con CAR dirigido a CD-19 con un dominio coestimulador de CD28 que usó un vector retrovírico para la transducción génica.
- Este producto de células T con CAR-CD19 indujo respuestas completas en el 70 % de los pacientes (14 de 20) (edad, 1–30 años) con LLA-B en recaída o resistente al tratamiento.
- La persistencia de las células T con CAR en este estudio fue de 1 a 2 meses; los pacientes que lograron RC presentaron recuperación de la linfocitopoyesis normal de células B.
- En un estudio de seguimiento posterior de 50 niños y adultos jóvenes que se trataron con células T con CAR-CD19, 21 pacientes lograron una RC con ERM negativa y recibieron un TCMH alogénico. En estos 21 pacientes, la mediana de SG fue de 70,2 meses y la tasa de SSC a 5 años fue del 61,9 %.[Nivel de evidencia C2]
- En otro informe se describió un ensayo multicéntrico de 25 niños y adultos jóvenes que recibieron tratamiento con células T con CAR anti-CD19 y anti-CD28z. Durante el ensayo, los investigadores aumentaron la dosis de ciclofosfamida linforreductora y analizaron los desenlaces sobre la base del acondicionamiento previo con dosis bajas y dosis altas, así como la presencia de ERM versus los indicios morfológicos de enfermedad antes del tratamiento.[Nivel de evidencia A1]
- Todos los efectos tóxicos fueron reversibles, incluso el síndrome de liberación de citocinas grave, que se presentó en un 16 % de los pacientes, y la neurotoxicidad grave, que se presentó en un 28 % de los pacientes.
- La tasa de respuesta completa general fue del 75 %, y el 89 % de los pacientes en RC fueron negativos para la ERM.
- La tasa de respuesta en la cohorte tratada con dosis alta de ciclofosfamida fue superior a la de la cohorte tratada con dosis baja (94 vs. 38 %); la tasa de respuesta fue más alta en la cohorte con ERM que en la cohorte con enfermedad morfológica (93 vs. 50 %).
- De los 18 pacientes que respondieron al tratamiento, 15 se sometieron a TCMH de consolidación.
- La mejora en la SG solo ocurrió en pacientes que recibieron acondicionamiento con dosis más altas de ciclofosfamida.
- En un esfuerzo por superar la pérdida temprana de células T con CAR dirigido a CD19 funcionales, se probó un abordaje 4-1BB humanizado en niños y adultos jóvenes con LLA-B o linfoma linfoblástico B recidivante o resistente al tratamiento. Los pacientes se trataron en 2 cohortes: 1) pacientes sin exposición previa a células T con CAR y 2) pacientes con exposición previa a CAR que no respondieron a los CAR más comunes, basados en anticuerpos derivados de ratón.
- El 98 % de los pacientes sin exposición previa a CAR y el 64 % de los pacientes con exposición previa lograron una RC.
- A los 6 meses, entre los pacientes que respondieron, el 27 % de aquellos sin exposición previa a CAR y el 48 % de aquellos con exposición previa perdieron la persistencia de CAR.
- En los pacientes sin exposición previa a CAR, la tasa de SSR fue del 84 % (IC 95 %, 72–97 %) a los 12 meses y del 74 % (IC 95 %, 60–90 %) a los 24 meses.
- En la cohorte sin exposición previa a CAR, la tasa de SSR fue del 74 % (IC 95 %, 56–97 %) a los 12 meses y del 58 % (IC 95 %, 37–90 %) a los 24 meses.
- Si bien estos resultados son favorables, no se compararon en forma directa con los de las células T con CAR que están aprobadas en la actualidad.
Terapia de células T con receptor de antígeno quimérico dirigido a CD19 para el tratamiento de la enfermedad en el sistema nervioso central y extramedular
Aunque existe la preocupación de que el tratamiento de pacientes con enfermedad en el sistema nervioso central (SNC) podría aumentar el riesgo de neurotoxicidad relacionada con la terapia de células T con receptor de antígeno quimérico (CAR), en estudios recientes se ha demostrado que los pacientes con enfermedad en el SNC que reciben este tipo de terapia tienen desenlaces similares a los de aquellos que no tienen enfermedad en el SNC, y no se observa aumento del síndrome de neurotoxicidad asociado a células inmunoefectoras (ICANS).
Evidencia (terapia de células T con CAR para pacientes con enfermedad en el SNC):
- Enfermedad activa en el SNC en el momento de la recaída o de la infusión.
- En un informe de un solo centro se analizaron 195 pacientes que recibieron de manera secuencial células T con CAR dirigido a CD19 4-1BB; 66 de estos pacientes tenían enfermedad en el SNC (43 con enfermedad aislada en el SNC y 23 con enfermedad combinada de médula ósea y SNC).
- En general, no hubo diferencias en las tasas de remisión ni en las tasas de supervivencia sin progresión con enfermedad en el SNC o sin esta.
- Los pacientes con compromiso aislado en el SNC tuvieron una tasa de SSR a 2 años del 66 % (IC 95 %, 52–82 %), en comparación con el 58 % (IC 95 %, 50–68%) en pacientes con compromiso de la médula ósea (P = 0,15).
- El 98 % de los pacientes se trataron por enfermedad en el SNC antes de la infusión y lograron grados SNC1 o SNC2 en el momento del tratamiento de células T con CAR. Los pacientes con enfermedad en el SNC sintomática o progresiva no recibieron tratamiento hasta que la enfermedad estuvo controlada y estable.
- En un informe retrospectivo se examinaron los desenlaces de 184 pacientes que se trataron con tisagenlecleucel en 15 instituciones de los Estados Unidos. En estas series, los pacientes se derivaron con el fin de recibir una infusión de células T con CAR para enfermedad solo medular (n = 129), en el SNC (n = 40; aislada [n = 23] o combinación de enfermedad medular y en el SNC [n = 17]) o enfermedad extramedular fuera del SNC (n = 15; 6 pacientes con enfermedad extramedular aislada y 9 pacientes con compromiso medular). Todos los pacientes, excepto uno con enfermedad en el SNC, recibieron tisagenlecleucel después de una o más recaídas (ninguno se había sometido a trasplante), mientras que 29 de los pacientes con enfermedad medular sola (22 %) recibieron infusiones en la primera RC, 10 de los cuales (7,8 %) se habían sometido a trasplante en el pasado.
- De los pacientes que se derivaron para recibir tisagenlecleucel por enfermedad en el SNC (n = 40), 31 (78 %) ya presentaban enfermedad negativa en el SNC en el momento de la infusion y el 88 % (35 de 40) estaban en RC el día 28 posterior a la infusión. De los 15 pacientes con enfermedad extramedular fuera del SNC, 10 (66 %) estaban en RC el día 28.
- La tasa de SSR a 12 meses fue del 59,4 % en los pacientes con enfermedad solo medular, del 59,4 % en los pacientes con enfermedad en el SNC (aislada o combinada) y del 50 % en aquellos con enfermedad extramedular fuera del SNC (aislada o combinada) (P = 0,92).
- En la cohorte de enfermedad en el SNC, no hubo diferencias significativas en la SSR a 12 meses entre los pacientes con enfermedad aislada en el SNC y aquellos con una combinación de enfermedad medular y en el SNC (P = 0,63).
- Se observaron efectos neurotóxicos de grado 3 o 4 en 3 de 40 pacientes (7,5 %) de la cohorte con enfermedad en el SNC, en comparación con 9 de los 129 pacientes (7 %) con enfermedad solo medular. Ninguno de los pacientes con enfermedad activa en el SNC en el momento de la infusión presentó efectos neurotóxicos de grado 3 o 4.
- En un segundo informe multicéntrico se revisaron los datos de 55 pacientes con recaídas que afectaban el SNC (aisladas o con compromiso de médula ósea) y que recibieron, en el ámbito institucional, células T con CAR dirigido a CD28 (n = 12) o tisagenlecleucel (vector 4-1BB, n = 43).
- Se logró la remisión en el 92 % de los niños que recibieron células T con CAR dirigido a CD28 y en el 93 % de los niños que recibieron células T con CAR dirigido a 4-1BB.
- De los 11 niños que recibieron células T con CAR dirigido a CD28 y alcanzaron una RC, 9 se sometieron a TCMH. Y 7 de estos 9 pacientes sobrevivieron sin recaída.
- Ninguno de los 40 pacientes que recibieron células T con CAR dirigido a 4-1BB (tisagenlecleucel) y alcanzaron una RC se sometió a un TCMH. De estos pacientes, 19 presentaron recaídas, 12 de las cuales afectaron el SNC.
- Los pacientes que recibieron tisagenlecleucel por una recaída aislada en el SNC tuvieron una incidencia alta de recaídas posteriores en el SNC (6 de 8 pacientes).
- En un informe de un solo centro se analizaron 195 pacientes que recibieron de manera secuencial células T con CAR dirigido a CD19 4-1BB; 66 de estos pacientes tenían enfermedad en el SNC (43 con enfermedad aislada en el SNC y 23 con enfermedad combinada de médula ósea y SNC).
- Los datos disponibles de pacientes con enfermedad extramedular que no afecta el SNC son escasos.
- En el informe de 195 pacientes que me mencionó anteriormente se comparó la enfermedad extramedular en el SNC, la enfermedad extramedular que no afecta el SNC y la enfermedad que afecta solo la médula ósea. No se encontraron diferencias entre los grupos en la supervivencia general ni en la SSR.
- Por el contrario, en un análisis retrospectivo de gran tamaño se observó que la enfermedad extramedular activa en el momento de la infusión se relacionaba de manera independiente con una SSC más precaria (CRI, 1,9; P = 0,01).
- En un análisis separado se encontró que, de 15 pacientes con enfermedad extramedular que no afectaba el SNC, 10 alcanzaron una RC y 6 de esos 10 pacientes presentaron una recaída posterior. Por consiguiente, solo 4 de 15 pacientes permanecieron sin complicaciones durante el periodo de seguimiento.
- En un análisis retrospectivo del NCI se identificaron limitaciones en la eliminación de la enfermedad extramedular que no afecta el SNC mediante la terapia de células T con CAR, a pesar de las respuestas simultáneas en la médula ósea, lo que destaca la función importante de la tomografía por emisión de positrones en el seguimiento.
Factores de riesgo relacionados con el fracaso de la terapia de células T con receptor de antígeno quimérico dirigido a CD19
En los ensayos iniciales en los que se usó la terapia de células T con receptor de antígeno quimérico (CAR) se demostró que una proporción muy alta de pacientes con enfermedad recidivante o resistente al tratamiento lograron RC, con independencia del recuento de glóbulos blancos, las características citogenéticas, el número de tratamientos previos, la respuesta a la quimioterapia u otros factores que tradicionalmente se relacionaron con la respuesta a la quimioterapia. En estudios posteriores se identificaron los siguientes factores relacionados con respuestas a largo plazo precarias en pacientes que lograron RC iniciales después de las infusiones de células T con CAR:
- Carga tumoral en el momento de la infusión de células T con CAR.
- En un estudio de un solo centro se evaluó el porcentaje de blastocitos en la médula ósea justo antes de la infusión de células T con CAR (después de la quimioterapia linfocitorreductora). La carga tumoral alta (CTA) se definió como aquella con un 40 % o más de blastocitos, y la carga tumoral baja (CTB) como aquella de menos de un 40 % de blastocitos.
- En la cohorte de pacientes con CTA (n = 15), la mejor tasa de respuesta general fue del 87 %, y la tasa de RC negativa para la ERM fue del 80 %.
- En la cohorte de pacientes con CTB (n = 55), la mejor tasa de respuesta general fue del 100 %, y todos los que presentaron respuesta fueron negativos para la ERM.
- En los pacientes con CTA, la probabilidad de continuar en remisión fue del 49 % (IC 95 %, 27–88 %) a los 12 meses y del 39 % (IC 95 %, 19–82 %) a los 24 meses.
- En los pacientes con CTB, la probabilidad de continuar en remisión fue del 86 % (IC 95 %, 77–96 %) a los 12 meses y del 78 % (IC 95 %, 67–91 %) a los 24 meses.
- En un estudio de 420 pacientes se evaluaron los factores de riesgo relacionados con la supervivencia a largo plazo.
- En el análisis multivariante, una carga de enfermedad del 5 % o más alta, calculada mediante citometría de flujo de la médula ósea antes de comenzar la quimioterapia linfocitorreductora, se relacionó con un cociente de riesgos instantáneos (CRI) de 2,52 (IC 95 %, 1,86–3,41) para la SSC inferior, en comparación con los pacientes que tenían una carga de enfermedad más baja.
- En un estudio de 180 pacientes que recibieron infusiones de tisagenlecleucel, en los análisis univariantes y multivariantes se observó que una carga de morbilidad alta (>5 % de blastocitos en la médula ósea, enfermedad SNC3 [definida como una muestra de líquido cefalorraquídeo con ≥5 glóbulos blancos/μl con linfoblastos o la presencia de parálisis de nervios craneales] o enfermedad extramedular [EEM] que no afecta el SNC) se relacionó con desenlaces inferiores.
- Los pacientes con una carga de enfermedad alta tuvieron desenlaces inferiores (tasa de SG a 12 meses, 58 %; tasa de SSC, 31 %) que los pacientes con carga de enfermedad baja (tasa de SG a 12 meses, 85 %; tasa de SSC, 70 %) y los pacientes con enfermedad indetectable (tasa de SG a 12 meses, 95 %; tasas de SSC, 72 %; P< 0,0001 para la SG y la SSC).
- En un estudio de un solo centro se evaluó el porcentaje de blastocitos en la médula ósea justo antes de la infusión de células T con CAR (después de la quimioterapia linfocitorreductora). La carga tumoral alta (CTA) se definió como aquella con un 40 % o más de blastocitos, y la carga tumoral baja (CTB) como aquella de menos de un 40 % de blastocitos.
- Fracaso previo de respuesta al blinatumomab.
- En un estudio de 420 pacientes pediátricos y adultos jóvenes con LLA que se trataron con células T con CAR dirigido a CD19, se notificaron los siguientes resultados:
- Los pacientes que en el pasado no habían presentado respuesta al tratamiento con blinatumomab antes de la terapia de células T con CAR tuvieron tasas de RC posteriores a la terapia de células T con CAR más bajas (20 de 31, 64,5 %), en comparación con aquellos que en el pasado habían respondido al blinatumomab (39 de 42, 92,9 %) o aquellos que nunca habían recibido blinatumomab (317 de 339, 93,5 %; P< 0,0001).
- Los que no habían respondido al blinatumomab tuvieron tasas de SSC a 6 meses más precarias (27,3 %; IC 95 %, 13,6–43,0 %) que los que habían respondido al blinatumomab (66,9%; IC 95 %, 50,6–78,9 %; P< 0,0001) o aquellos que nunca habían recibido blinatumomab (72,6 %; IC 95 %, 67,5–77 %; P< 0,0001).
- En un estudio de 420 pacientes pediátricos y adultos jóvenes con LLA que se trataron con células T con CAR dirigido a CD19, se notificaron los siguientes resultados:
- Aplasia de células B persistente y secuenciación de próxima generación de ERM (NGS-ERM).
- En estudios preliminares se determinó que la aplasia de células B era una medida funcional de la persistencia de células T con CAR dirigido a CD19, y que la pérdida de aplasia de células B durante los primeros meses de tratamiento producía tasas altas de recaída. Sin embargo, esta es una medida imperfecta ya que puede presentarse una recaída negativa para CD19 a pesar de la persistencia de aplasia de células B. Las técnicas de NGS-ERM se basan en reordenamientos clonales en la IgH o secuencias TCR en linfoblastos que, por lo general, persisten aunque haya pérdida de expresión de CD19 de superficie.
- Para definir el riesgo de recaída, en un estudio se evaluó la aplasia de células B y la NGS-ERM en la médula ósea a lo largo del tiempo en 143 pacientes que recibieron tisagenlecleucel.
- En un análisis multivariante en el día 28 se observaron relaciones independientes entre la NGS-ERM superior a 0 (CRI, 4,87; IC 95 %, 2,18–10,8; P< 0,001) y la pérdida de aplasia de células B (CRI, 3,33; IC 95 %, 1,44–7,69; P = 0,005) con la recaída.
- A los 3 meses, el CRI de NGS-ERM aumentó a 12 (IC 95 %, 2,87–50; P< 0,001), mientras que la pérdida de aplasia de células B no fue predictiva de manera independiente (CRI, 1,27; IC 95 %, 0,33–4,79; P = 0,7).
- La pérdida temprana de aplasia de células B predijo mejor una recaída que la pérdida tardía de aplasia de células B.
- La pérdida de aplasia de células B en los primeros 6 meses produjo una tasa de SSC inferior al 20 %; sin embargo, los pacientes que presentaron pérdida de aplasia de células B a 1 año, tuvieron una tasa de SSC del 75 %.
- Cabe destacar que una NGS-ERM positiva en cualquier momento después de la terapia de células T con CAR produjo un riesgo de recaída muy alto.
- Dosis de células T con CAR.
- En un estudio multicéntrico retrospectivo se revisaron las dosis de células T con CAR dirigido a CD19 (tisagenlecleucel) administradas por kilogramo por cuartiles y se evaluaron los desenlaces de los pacientes.
- Los pacientes que recibieron la dosis más alta por cuartil presentaron SG (CRI, 0,22; IC 95 %, 0,08–0,61), SSC (CRI, 0,24; IC 95 %, 0,12–0,49) y SSR (CRI, 0,18; IC 95 %, 0,07–0,49) superiores, en comparación con los que recibieron la dosis más baja por cuartil.
- Las dosis más altas de células T con CAR no se relacionaron con efectos tóxicos adicionales.
- Un posible factor de confusión fue el número significativamente más alto de pacientes con características citogenéticas favorables que recibieron la dosis más alta por cuartil.
- Se necesita más investigación para determinar la relación entre el umbral de dosificación y los desenlaces posteriores a la terapia de células T con CAR en la que se usó tisagenlecleucel.
- En un estudio multicéntrico retrospectivo se revisaron las dosis de células T con CAR dirigido a CD19 (tisagenlecleucel) administradas por kilogramo por cuartiles y se evaluaron los desenlaces de los pacientes.
- Momento de la recaída después del TCMH y antes del tratamiento de células T con CAR dirigido a CD19.
- En un estudio retrospectivo se notificaron los desenlaces de 81 pacientes con recaídas múltiples o LLA-B primaria resistente al tratamiento que recibieron tisagenlecleucel. De este grupo pacientes, el 80 % se había sometido a TCMH alogénico.
- En el día 28 de la terapia de células T con CAR, 71 de 81 pacientes (87,7 %) estaban en remisión.
- Después de 2 años de la terapia de células T con CAR, la tasa de SSC fue del 45,3 %, y la tasa de SG fue del 53,2 %.
- En los pacientes que se habían sometido a trasplante, el tiempo hasta la recidiva después del TCMH fue un factor predictor significativo del desenlace al recibir tisagenlecleucel. Los pacientes que recayeron en el transcurso de 6 meses desde el TCMH presentaron una tasa de SSC del 18,4 %, en comparación con el 55,5 % de aquellos que recayeron a los 6 meses o después del TCMH.
- En un estudio retrospectivo se notificaron los desenlaces de 81 pacientes con recaídas múltiples o LLA-B primaria resistente al tratamiento que recibieron tisagenlecleucel. De este grupo pacientes, el 80 % se había sometido a TCMH alogénico.
Recaída o falta de respuesta a la terapia de células T con receptor de antígeno quimérico dirigido a CD19
En estudios retrospectivos se han descrito desenlaces y evaluado factores relacionados con la supervivencia después de una recaída en pacientes que recibieron terapia de células T con receptor de antígeno quimérico (CAR) dirigido a CD19:
- En un estudio se evaluaron 166 recaídas que se presentaron después de la terapia de células T con CAR en la que se utilizaron los dominios coestimuladores 4-1BB o CD28.
- De los pacientes, 83 (50 %) eran positivos para CD19, 68 (41 %) eran negativos para CD19 y 12 (7,2 %) presentaron recaídas con cambio de linaje.
- La mediana de SG fue de 11,9 meses (IC 95 %, 9–17 meses), sin diferencias entre las recaídas positivas para CD19 y las negativas para CD19.
- Los pacientes que presentaron recaídas con cambio de linaje tuvieron un pronóstico desalentador, y no hubo sobrevivientes a largo plazo.
- Las recaídas positivas para CD19 se relacionaron con una mayor carga de tratamiento previo; esto se determinó por la presencia de un mayor número de remisiones previas.
- Las recaídas negativas para CD19 se relacionaron con una mayor carga de enfermedad antes de la infusión de células T con CAR, falta de respuesta previa a blinatumomab, edad avanzada y dominio coestimulador 4-1BB.
- Las recaídas con cambio de linaje se relacionaron con la presencia de reordenamientos de KMT2A.
- En un segundo estudio se revisaron 80 pacientes (27 que no respondieron y 53 que recayeron) que recibieron terapia de células T con CAR dirigido a CD19 y dominio 4-1BB, y que no lograron remisiones.
- Entre los pacientes que no respondieron al tratamiento, el 95 % presentó una carga de enfermedad elevada (blastocitos, >5 %) antes de la infusión de células T con CAR.
- Las recaídas negativas para CD19 se relacionaron con una disminución de la tasa de SG a 1 año, en comparación con las recaídas positivas para CD19 (30 % [IC 95 %, 14–66 %] vs. 68 % [49–92%]; P = 0,0068).
- Solo el tratamiento seguido de TCMH, en el caso de lograr una remisión, se relacionó con una supervivencia a largo plazo.
Terapia anterior de células T con receptor de antígeno quimérico dirigido a CD19 derivado de donante alogénico
Es posible que ciertos pacientes no sean susceptibles de producir células T con receptor de antígeno quimérico (CAR) autógeno dirigido a CD19, como aquellos que presentan una recaída muy temprana después de un TCMH. La terapia de células T con CAR que utiliza células producidas a partir del donante de un paciente determinado que ha recibido un TCMH puede ser una opción para estos pacientes. Un pequeño grupo de pacientes que recayeron después de un TCMH recibieron células T con CAR dirigido a CD19 alogénicas que se produjeron a partir de células T obtenidas de donantes. Los 13 pacientes lograron una RC de médula ósea negativa para la ERM, con una RC del 83 % en sitios extramedulares. De los pacientes, 3 se sometieron a un TCMH posterior. Al cabo de una mediana de seguimiento de 12 meses, 8 pacientes permanecieron con una RC de médula ósea. Hubo solo 1 caso de EICH grave. Estos resultados proporcionan la evidencia preliminar para la inocuidad y la eficacia del uso de células T con CAR producidas a partir del donante alogénico anterior en el entorno de una recaída de la LLA.
Función del trasplante de células madre hematopoyéticas de consolidación después de la terapia de células T con receptor de antígeno quimérico dirigido a CD19 para los pacientes con leucemia linfoblástica aguda
En los estudios de abordajes de células T con receptor de antígeno quimérico (CAR) dirigido a CD19 de segunda generación se ha demostrado que los sistemas que utilizan moléculas coestimuladoras por medio de CD28 producen una vida media de las células T con CAR relativamente corta. Esta vida media corta conduce a tasas muy altas de recaída, a menos que se haga un TCMH poco después de la recuperación de los efectos tóxicos que produce la terapia de células T con CAR. Por lo tanto, las terapias de células T con CAR dirigido a CD28 se consideran tratamientos puente, y el TCMH para los pacientes aptos en general se planifica entre 4 y 8 semanas después del procedimiento de células T con CAR. Se ha demostrado que el tisagenlecleucel y otros CAR con moléculas coestimuladoras de 4-1BB presentan índices significativos de persistencia, lo que conduce a una remisión a largo plazo en el 45 % al 50 % de los pacientes sin terapia adicional. Hasta un 80 % de las recaídas ocurrieron durante el primer año posterior a la terapia de células T con CAR y hay una ventana de remisión profunda. Debido a este hallazgo, algunos grupos de estudio han defendido que se planifique el TCMH durante la remisión justo después de la infusión de células T con CAR, en todos los pacientes o en aquellos que no hayan tenido un TCMH previo. Este tema no se ha analizado en ensayos aleatorizados, pero algunos estudios han evaluado el tema de forma retrospectiva.
Evidencia (TCMH de consolidación después de la terapia de células T con CAR):
- Abordaje mediante terapia puente de células T con CAR CD28.
- En un estudio se evaluaron 50 niños y adultos jóvenes que se trataron con terapia de células T con CAR CD19 basada en CD28.[Nivel de evidencia C2]
- De los pacientes, 21 alcanzaron una RC negativa para la ERM y se sometieron a TCMH alogénico. En estos 21 pacientes, la mediana de SG fue de 70,2 meses y la tasa de SSC a 5 años fue del 61,9 %.
- En un estudio se evaluaron 50 niños y adultos jóvenes que se trataron con terapia de células T con CAR CD19 basada en CD28.[Nivel de evidencia C2]
- CAR 4-1BB con abordajes diferentes para el TCMH.
- En un estudio de 50 pacientes aptos para TCMH se notificaron los siguientes resultados después de la terapia de células T con CAR basada en 4-1BB (ensayo PLAT-02 [NCT02028455]):
- Los pacientes que perdieron la capacidad funcional de las células T con CAR en los 63 días posteriores a la terapia presentaron una SSL superior cuando se sometieron a un TCMH, en comparación con aquellos que no se sometieron a un TCMH (P = 0,01).
- Los pacientes sin antecedentes de TCMH tuvieron una SSL superior cuando se planificó el TCMH, con una tendencia hacia la significancia estadística (P = 0,09). Este beneficio no se observó en los 34 pacientes con antecedentes de TCMH (P = 0,45).
- En un estudio de 50 pacientes aptos para TCMH se notificaron los siguientes resultados después de la terapia de células T con CAR basada en 4-1BB (ensayo PLAT-02 [NCT02028455]):
- CAR 4-1BB seguido de un intento de TCMH.
- En un estudio realizado en China de 52 niños que recibieron terapia de células T con CAR dirigido a CD19 o CD22 y se sometieron a un TCMH planificado en una mediana de 50 días después de la infusión de células T con CAR (intervalo, 34–98 días; 48 pacientes con ERM y 4 pacientes positivos para la ERM), se notificaron los siguientes resultados:
- La tasa de SSC a 1 año fue del 73 % y la tasa de SG a 1 año fue del 87,7 %.
- A pesar de que la tasa de supervivencia de los pacientes aptos para un TCMH en remisión fue alta, en el análisis no se incluyeron los pacientes que habían recibido un TCMH con anterioridad, los pacientes que no alcanzaron la remisión con las infusiones de células T con CAR ni los pacientes que no resultaron aptos para el TCMH después de la terapia de células T con CAR.
- En un estudio realizado en China de 52 niños que recibieron terapia de células T con CAR dirigido a CD19 o CD22 y se sometieron a un TCMH planificado en una mediana de 50 días después de la infusión de células T con CAR (intervalo, 34–98 días; 48 pacientes con ERM y 4 pacientes positivos para la ERM), se notificaron los siguientes resultados:
Terapia de células T con receptor de antígeno quimérico dirigido a CD22
Por lo menos el 50 % de las recaídas después de la terapia de células T con receptor de antígeno quimérico (CAR) dirigido a CD19 se presentaron por escape antigénico, que se relaciona con las variantes de la proteína CD19 que eliminan los sitios de unión utilizados por los conjuntos de células T con CAR. El rescate después del escape antigénico se documentó con abordajes de terapia celular e inmunitaria dirigidos a un antígeno linfoide secundario, el CD22. No se han publicado estudios de evaluación específica del rescate con inotuzumab en el caso de recaída negativa para CD19, pero 2 grupos notificaron tasas altas de remisión y supervivencia posteriores, por lo general, cuando a la terapia de células T con CAR CD22 le siguió un TCMH.; [Nivel de evidencia C1]; [Nivel de evidencia C2] Debido a que se puede reducir la expresión del antígeno CD22, hay preocupación sobre las consecuencias de usar CD22 como la única diana molecular en la respuesta a largo plazo a las células T con CAR; por consiguiente, este abordaje a menudo se acompaña de un TCMH.
Evidencia (terapia de células T con CAR dirigido a CD22):
- Los investigadores del NCI notificaron un ensayo de fase I/II con 58 niños y adultos jóvenes tratados con un abordaje de células T con CAR dirigido a CD22.[Nivel de evidencia C2]
- De los 55 pacientes con LLA que recibieron la infusión, 40 alcanzaron RC (73 %); el 88 % de esos pacientes alcanzaron remisiones negativas para la ERM.
- A pesar de que el 86 % de los pacientes presentó síndrome de liberación de citocinas, en el 90 % de los casos fue de grado 1 a 2.
- El 33 % de los pacientes presentaron efectos neurotóxicos, todos ellos de grados 1 o 2, a excepción de 1 paciente, que tuvo una hemorragia intracraneal.
- El 38 % de los pacientes presentó un síndrome similar a la linfohistiocitosis hemofagocítica o síndrome de activación de macrófagos que, en ocasiones, requirió tratamiento con anakinra.
- Los pacientes que recibieron terapia dirigida a CD22 con inotuzumab o terapia de células T con CAR CD22 antes de esta terapia tuvieron tasas más bajas de expresión de CD22, menos probabilidades de alcanzar la remisión y una remisión más corta.
- De los 40 pacientes que alcanzaron RC, 30 (75 %) recayeron; solo los pacientes que se sometieron a TCMH tuvieron remisiones a largo plazo (14 pacientes se sometieron a TCMH, 6 recayeron). La mediana de SSR fue de 6 meses en este grupo de pacientes que alcanzó la RC.
- Un grupo chino administró células T con CAR dirigido a CD22 a 34 pacientes que no respondieron a la terapia de células T con CAR dirigido a CD19.
- De los pacientes evaluables en el día 30, el 80 % presentó una RC (71 % de todos los pacientes).
- No se administraron nuevos tratamientos en 7 pacientes, y 3 pacientes permanecieron en remisión 5 a 13 meses después de la terapia.
- De los pacientes, 11 recibieron un TCMH; la tasa de SSL a 1 año fue del 72 %.
- En este estudio se demostró que el rescate a largo plazo de los pacientes que no responden a las células T con CAR dirigido a CD19 se puede lograr usando células T con CAR dirigido a CD22 y TCMH.
Terapia de células T con receptor de antígeno quimérico dirigido a múltiples antígenos
Los investigadores han evaluado abordajes dirigidos a varios antígenos para el tratamiento de la LLA con el fin de solucionar la recaída causada por escape inmunológico. Los estudios han incluido los siguientes abordajes:
- Empleo de infusión simultánea o secuencial de 2 tipos de células T con receptor de antígeno quimérico (CAR) dirigido a diferentes antígenos y elaboradas de manera independiente.
- Un solo producto de células T con CAR elaboradas por un solo fabricante que contiene 2 poblaciones celulares, cada una dirigida a un antígeno diferente.
- Un solo producto que contiene un CAR con dominios para múltiples antígenos.
Si bien ninguno de estos abordajes está disponible para la compra, están en curso estudios con estos abordajes.
En un gran estudio en la China se incluyó a pacientes que recibieron una mezcla 1:1 de células T elaboradas de manera independiente con CAR dirigido a CD19 y CD22. Este tratamiento produjo la remisión en el 99 % de los pacientes. La persistencia de aplasia de células B después de 6 meses y el TCMH después de recibir las células T con CAR se asociaron, cada uno, con mejora de la supervivencia.
En otro estudio de China, se infundieron células T con CAR dirigido a CD19 y luego células T dirigidas a CD22 después de obtener la remisión. Esto conllevó una tasa de SSC a 18 meses del 80 %, y TCMH solo en el 10 % de los pacientes.
En otros 3 ensayos se evaluó un único proceso de elaboración de células T con CAR multidirigidos. El tratamiento con este tipo de células T con CAR produjo tasas de remisión razonables. Sin embargo, la duración de las células CAR T fue deficiente, y los desenlaces de los pacientes no fueron mejores con estas células T con CAR en comparación con las células T con CAR dirigido a CD19 que se encuentran disponibles para la compra.
Tratamiento de la recaída extramedular aislada
Con las mejoras en el tratamiento en los niños con LLA, la incidencia de recaída extramedular aislada ha disminuido. La incidencia de recaída aislada en el SNC es menor del 5 % y la recaída testicular es menor del 1 % al 2 %. Al igual que las recaídas en la médula ósea y las recaídas mixtas, el tiempo desde el diagnóstico inicial hasta la recaída es un factor pronóstico clave para las recaídas extramedulares aisladas. Además, en un estudio se observó que la edad superior a 6 años en el momento de la recaída fue un factor de pronóstico adverso en los pacientes con recaída extramedular aislada, mientras que en otros estudios se indicó que la edad de 10 años era un mejor límite. Cabe mencionar que, en la mayoría de los niños con recaídas extramedulares aisladas, se logra demostrar una enfermedad submicroscópica en la médula cuando se emplean técnicas moleculares sensibles y las estrategias de tratamiento exitosas deben ser eficaces para el control de la enfermedad local y sistémica. Los pacientes con una recaída aislada en el SNC que presentan una ERM superior al 0,01 % en una médula morfológicamente normal tienen un pronóstico más precario (tasa de SSC a 5 años, 30 %) que los pacientes sin ERM o que tienen una ERM inferior al 0,01 % (tasa de SSC a 5 años, 60 %).
Recaída aislada en el sistema nervioso central
Las opciones de tratamiento estándar para la LLA con recaída en el SNC son las siguientes:
- Quimioterapia sistémica e intratecal.
- Radiación craneal o craneoespinal.
- TCMH.
- Terapia de células T con CAR para la enfermedad aislada en el SNC con recaídas múltiples.
Si bien el pronóstico de los niños con recaída aislada en el SNC ha sido bastante precario en el pasado, el pronóstico ha mejorado con el uso de tratamiento sistémico o intratecal intensivo, seguido de radiación craneal o craneoespinal, sobre todo en los pacientes que no recibieron radiación craneal durante su primera remisión.
Evidencia (quimioterapia y radioterapia):
- En un estudio del Pediatric Oncology Group (POG), en el que se utilizó esta estrategia, los niños que no habían recibido radioterapia previa y cuya remisión inicial fue de 18 meses o más tuvieron una tasa de SSC a 4 años de cerca del 80 %, en comparación con las tasas de SSC de cerca del 45 % en los niños con recaída en el SNC en los 18 meses a partir del diagnóstico.
- En un estudio de seguimiento del POG, los niños que no habían recibido radioterapia previa y que tuvieron una remisión inicial de 18 meses o más se trataron con quimioterapia sistémica e intratecal intensiva durante 1 año seguida de 18 Gy de radioterapia craneal sola.
- La tasa de SSC a 4 años fue del 78 %. Los niños con una remisión inicial de menos de 18 meses también recibieron la misma quimioterapia, pero se sometieron a radiación craneoespinal (24 Gy craneal o 15 Gy espinal) como en el primer estudio del POG y lograron una SSC a 4 años del 52 %.
- En el estudio AALL02P2 (NCT00096135) del Children’s Oncology Group, se inscribieron 118 niños aptos con LLA-B y recaída aislada tardía en el SNC. Los pacientes recibieron tratamiento sistémico intensificado, quimioterapia intratecal triple y 12 Gy de irradiación craneal administrada a los 12 meses; la quimioterapia de mantenimiento continuó hasta 104 semanas después del diagnóstico.
- La tasa de SSC a 3 años fue del 64,3 % (± 4,5 %) y la tasa de SG fue del 79,6 % (± 3,8%).
- Se presentaron segundas recaídas que incluían el SNC en el 46,1 % de los pacientes (18 de 39).
- De los 112 pacientes que completaron el tratamiento, 78 recibieron la radioterapia especificada en el protocolo.
- La inscripción de pacientes en el estudio se interrumpió después de que el análisis de seguimiento provisional demostró una SSC inferior, en comparación con el estudio POG 9412, en el que los pacientes recibieron 18 Gy de radiación craneal. Por lo tanto, una dosis más baja de radiación craneal se consideró inadecuada.
Se publicaron varias series de casos que describen el TCMH para tratar la recaída aislada en el SNC. Aunque en algunos informes se indicó una posible función del TCMH para los pacientes con enfermedad aislada en el SNC con recaída muy temprana y enfermedad de células T, la evidencia es menor sobre la necesidad de un TCMH en el momento de una recaída temprana aislada en el SNC con enfermedad de células B y no hay evidencia de la utilidad en la recaída tardía. En el estudio COG AALL0433, los pacientes con LLA-B y una recaída aislada en el SNC muy temprana se trataron con quimioterapia intensiva y radiación craneal o TCMH alogénico después de que se alcanzara la segunda RC, según la disponibilidad de donantes y de la decisión de los médicos. Un pequeño número de pacientes se sometió a TCMH (n = 7), lo que se relacionó con una SSE y una SG más favorables, en comparación con los pacientes que continuaron con quimioterapia y radioterapia.
Evidencia (trasplante de células madre hematopoyéticas):
- En otro estudio, se comparó el desenlace de los pacientes tratados con trasplantes de hermanos con compatibilidad de HLA o quimiorradioterapia, como en los estudios del POG mencionados antes.[Nivel de evidencia C2] En este estudio se incluyeron trasplantes realizados durante la recaída temprana (<18 meses a partir del diagnóstico) y la recaída tardía.
- La probabilidad a 8 años ajustada por la edad de SSL (58 %) y la duración de la primera remisión (66 %) fueron semejantes.
- Debido al resultado relativamente favorable de los pacientes con recaída aislada en el SNC más de 18 meses después del diagnóstico que se trataron con quimiorradioterapia sola (>75 %), el COG por lo general no recomienda el trasplante para este grupo.
- En el ensayo MRC ALLR3 se probó la inducción intensiva con mitoxantrona versus idarrubicina en pacientes con LLA recidivante y se estableció que el desenlace es superior cuando se usó mitoxantrona. En el subanálisis de 80 pacientes que entraron al ensayo con recaída aislada del SNC, se incluyeron 13 pacientes con recaída muy temprana (definida como <18 meses desde el diagnóstico inicial), 55 pacientes con recaída temprana (definida como >18 meses desde el diagnóstico inicial, pero durante los 6 meses siguientes al tratamiento) y 12 pacientes con recaída tardía.[Nivel de evidencia B4]
- Los pacientes con recaída tardía evolucionaron mejor con quimioterapia y radioterapia craneal y 11 de 12 pacientes sobrevivieron.
- El TCMH alogénico se recomendó para la recaída muy temprana y temprana. Después de los 3 cursos de inducción planificados, 66 pacientes estaban vivos y no presentaban recaída. Fueron aptos para el TCMH recomendado por el protocolo 54 pacientes con recaída aislada del SNC temprana y muy temprana; 39 (72 %) pacientes recibieron un TCMH. El 21 % de esos pacientes presentaron recaída, en comparación con una tasa de recaída del 71 % en el grupo que no recibió TCMH.
- De los pacientes aptos para trasplante, la terapia con mitoxantrona durante la reinducción, en lugar de la terapia con idarrubicina, se relacionó con una ventaja de supervivencia (tasas de supervivencia sin progresión a 3 años, 61 vs. 21 %; P = 0,027). De la misma manera que en el estudio más grande, la ventaja más importante del grupo de mitoxantrona se presentó en quienes recibieron un TCMH. El número bajo de pacientes del grupo de recaída muy temprana impidió el análisis detallado de esta cohorte; las tasas de fracaso en el grupo de recaída temprana que recibió quimioterapia y radioterapia craneal fueron inferiores a las otras publicadas, lo cual pone en tela de juicio este abordaje de quimioterapia para los pacientes con recaída temprana aislada del SNC.
Evidencia (terapia de células T con CAR para la enfermedad aislada en el SNC con recaídas múltiples):
Se ha demostrado que las células CAR T penetran en el SNC y producen tasas altas de remisión en pacientes con enfermedad en el SNC con compromiso de la médula ósea o sin este. Una pequeña cantidad de estudios han abordado la relación del compromiso del SNC con los resultados de la terapia de células T con CAR.
- En un estudio de un solo centro se analizaron 195 pacientes secuenciales tratados con células T con CAR 4-1BB dirigidas a CD19, y 66 de estos pacientes tenían enfermedad en el SNC (43 con enfermedad aislada en el SNC, 23 con enfermedad combinada de médula ósea y SNC).
- En general, no hubo diferencias en las tasas de remisión y en la supervivencia sin progresión en los pacientes con enfermedad en el SNC y sin esta.
- De los pacientes con enfermedad en el SNC, el 79 % presentaba una segunda recaída o superior.
- Los pacientes con compromiso del SNC aislado tuvieron una tasa de SSR a 2 años del 66 % (IC 95 %, 52–82 %), en comparación con el 58 % (IC 95 %, 50–68%) en pacientes con compromiso de la médula ósea (P = 0,15).
Recaída testicular aislada
Los resultados del tratamiento de la recaída testicular aislada dependen del momento de la recaída. La tasa de SSC a 3 años es de alrededor del 40 % en los niños con recaída testicular evidente durante la terapia y de alrededor del 85 % para los niños con recaída testicular tardía.
Las opciones de tratamiento estándar en América del Norte para la LLA infantil con recaída testicular son las siguientes:
- Quimioterapia.
- Radioterapia.
Los abordajes estándar para el tratamiento de la recaída testicular aislada en América del Norte incluyen radioterapia local con quimioterapia intensiva. En algunos grupos de ensayos clínicos europeos se realiza una orquiectomía del testículo comprometido en lugar de administrar radiación. Se obtiene una biopsia del otro testículo en el momento de la recaída para determinar si se debe realizar control local adicional (extirpación quirúrgica o radiación). En un estudio, en el que se obtuvieron biopsias testiculares al final del tratamiento de primera línea, no se logró demostrar ningún beneficio de la supervivencia para los pacientes con detección temprana de la enfermedad oculta.
Hay pocos datos clínicos de desenlaces que no incluyan el uso de radioterapia u orquiectomía. Los protocolos de tratamiento en los que se probó este abordaje incorporaron dosis intensificadas de quimioterapéuticos (por ejemplo, dosis altas de metotrexato) que tal vez produzcan concentraciones antileucémicas en los testículos.
Evidencia (tratamiento de la recaída testicular):
- En el ensayo COG AALL02P2 (NCT00096135) se probó si se podía omitir el uso de radioterapia en los pacientes con recaída testicular aislada (más de 18 meses después del diagnóstico). En este ensayo, el tamaño testicular se reevaluó después del primer mes de quimioterapia de reinducción, que incluyó dosis altas de metotrexato. Si el testículo continuaba agrandado, se procedía con una biopsia y si el resultado era de compromiso entonces los pacientes recibían radioterapia local. Los pacientes que recuperaban el tamaño testicular o que no tenían compromiso en la biopsia se trataron sin usar radioterapia. La quimioterapia posinducción para todos los pacientes (con irradiación previa o sin esta) incluyó múltiples cursos de dosis altas de metotrexato.
- De 40 pacientes inscritos, 26 tuvieron un agrandamiento testicular persistente después de la reinducción. De esos 26 pacientes, 12 presentaron biopsia testicular positiva para el cáncer y 11 de ellos recibieron radioterapia testicular; el resto de los pacientes del ensayo se trataron sin radiación.
- Los participantes que recibieron radioterapia testicular alcanzaron una tasa de SSC a 5 años del 73 % versus el 61 % en aquellos que no recibieron radioterapia (P = 0,6); la tasa de SG a 5 años fue del 73 % en los que recibieron radioterapia testicular versus el 71 % (P = 0,9) en aquellos que no recibieron radioterapia testicular.
- Por lo tanto, es viable omitir la radioterapia en los pacientes con recaída testicular aislada que lograron respuesta favorable después de la inducción inicial (documentada por reducción del tamaño o biopsia).
- En los Países Bajos, los investigadores trataron a 5 niños con recaída testicular tardía con dosis altas de metotrexato durante la inducción (12 g/m2) y en intervalos regulares durante el resto del tratamiento (6 g/m2) sin radioterapia testicular.
- Los 5 niños fueron sobrevivientes a largo plazo.
- En una serie pequeña de niños que habían presentado recaída testicular aislada después de un TCMH por una recaída sistémica previa de LLA, 5 de 7 niños presentaron SSC prolongada sin un segundo TCMH.[Nivel de evidencia C1]
Opciones de tratamiento en evaluación clínica para la leucemia linfoblástica aguda infantil en recaída
Ensayos para la leucemia linfoblástica aguda durante la primera recaída
La información en inglés sobre los ensayos clínicos patrocinados por el NCI se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presentan ejemplos de ensayos clínicos nacionales o institucionales en curso:
- COG-AALL1821 (NCT04546399) (A Study to Compare Blinatumomab Alone to Blinatumomab With Nivolumab in Patients Diagnosed With First Relapse B-ALL): En este protocolo se está probando la incorporación de inmunoterapia en el tratamiento de pacientes con LLA-B que presentan una primera recaída medular. Los pacientes se estratifican en 3 grupos de acuerdo a la edad cuando ingresan al estudio y el momento de la recaída desde el diagnóstico inicial.
- El grupo 1 incluye los pacientes que cumplen una de las siguientes características: (1) mayor de 18 años (independientemente del momento de la recaída) o (2) menor de 18 años que presenta recaída antes de los 24 meses desde el diagnóstico inicial. Estos pacientes se asignan al azar para recibir blinatumomab solo o en combinación con nivolumab (inhibidor de puntos de control), como reinducción durante 1 o 2 ciclos, seguido de TCMH alogénico.
Todos los pacientes que no pertenecen al grupo 1 (es decir, aquellos menores de 18 años con una recaída que se presenta más de 24 meses después del diagnóstico inicial) reciben una reinducción con 4 fármacos (vincristina, dexametasona, doxorrubicina y pegaspargasa) y luego se clasifican como de grupo 2 o grupo 3.
- Los pacientes del grupo 2 son menores de 18 años con una de las siguientes características: (1) recaída que ocurre entre los 24 meses y los 36 meses desde el diagnóstico, independientemente de la ERM al final de la reinducción o (2) recaída que se presenta más de 36 meses después del diagnóstico con ERM superior al 0,1 % después de la reinducción con 4 fármacos. Estos pacientes se asignan al azar para recibir blinatumomab solo o en combinación con nivolumab, como consolidación posinducción, antes de someterse a TCMH alogénico.
- Los pacientes del grupo 3 son aquellos menores de 18 años que presentan una recaída después de los 36 meses del diagnóstico y una ERM inferior al 0,1 % después de la reinducción con 4 fármacos y que no se someten a TCMH. Estos pacientes se asignan al azar para recibir quimioterapia de consolidación estándar que se alterna con 3 ciclos de monoterapia con blinatumomab o blinatumomab en combinación con nivolumab, seguida de terapia de mantenimiento.
Los pacientes con recaída extramedular aislada no son aptos para este ensayo.
- TACL 2012-002 (NCT02879643) (Vincristine Sulfate Liposome Injection in Combination with UKALL R3 Induction Chemotherapy for Children, Adolescents, and Young Adults with Relapsed ALL): en este ensayo se evalúa la inocuidad y la viabilidad del uso de inyecciones de sulfato de vincristina liposomal en reemplazo de la vincristina estándar para el régimen de inducción de UKALL R3 en los pacientes con LLA (LLA-B o LLA-T) en el momento de la primera, segunda o tercera recaída. Son aptos para participar los pacientes con compromiso medular M2 (5–24 % blastocitos) o M3 (>25 % blastocitos).
Ensayos para la leucemia linfoblástica aguda en segunda y posteriores recaídas, o resistente al tratamiento
Se dispone de varios ensayos clínicos en los que se investigan nuevos fármacos y combinaciones de fármacos para niños con LLA en segunda o posteriores recaídas o resistente al tratamiento. En estos ensayos se examinan los tratamientos dirigidos específicos para la LLA, como las terapias a base de anticuerpos monoclonales y fármacos que inhiben las vías de transducción de señales necesarias para la proliferación y la supervivencia de las células leucémicas. Están en investigación nuevos fármacos, combinaciones de fármacos y abordajes inmunoterapéuticos en varios ensayos clínicos. Para obtener más información en inglés, consultar el portal de Internet ClinicalTrials.gov.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
References
- Reismüller B, Attarbaschi A, Peters C, et al.: Long-term outcome of initially homogenously treated and relapsed childhood acute lymphoblastic leukaemia in Austria--a population-based report of the Austrian Berlin-Frankfurt-Münster (BFM) Study Group. Br J Haematol 144 (4): 559-70, 2009.
- Uderzo C, Conter V, Dini G, et al.: Treatment of childhood acute lymphoblastic leukemia after the first relapse: curative strategies. Haematologica 86 (1): 1-7, 2001.
- Chessells JM, Veys P, Kempski H, et al.: Long-term follow-up of relapsed childhood acute lymphoblastic leukaemia. Br J Haematol 123 (3): 396-405, 2003.
- Rivera GK, Zhou Y, Hancock ML, et al.: Bone marrow recurrence after initial intensive treatment for childhood acute lymphoblastic leukemia. Cancer 103 (2): 368-76, 2005.
- Einsiedel HG, von Stackelberg A, Hartmann R, et al.: Long-term outcome in children with relapsed ALL by risk-stratified salvage therapy: results of trial acute lymphoblastic leukemia-relapse study of the Berlin-Frankfurt-Münster Group 87. J Clin Oncol 23 (31): 7942-50, 2005.
- Schroeder H, Garwicz S, Kristinsson J, et al.: Outcome after first relapse in children with acute lymphoblastic leukemia: a population-based study of 315 patients from the Nordic Society of Pediatric Hematology and Oncology (NOPHO). Med Pediatr Oncol 25 (5): 372-8, 1995.
- Wheeler K, Richards S, Bailey C, et al.: Comparison of bone marrow transplant and chemotherapy for relapsed childhood acute lymphoblastic leukaemia: the MRC UKALL X experience. Medical Research Council Working Party on Childhood Leukaemia. Br J Haematol 101 (1): 94-103, 1998.
- Buchanan GR, Rivera GK, Pollock BH, et al.: Alternating drug pairs with or without periodic reinduction in children with acute lymphoblastic leukemia in second bone marrow remission: a Pediatric Oncology Group Study. Cancer 88 (5): 1166-74, 2000.
- Rivera GK, Hudson MM, Liu Q, et al.: Effectiveness of intensified rotational combination chemotherapy for late hematologic relapse of childhood acute lymphoblastic leukemia. Blood 88 (3): 831-7, 1996.
- Bührer C, Hartmann R, Fengler R, et al.: Peripheral blast counts at diagnosis of late isolated bone marrow relapse of childhood acute lymphoblastic leukemia predict response to salvage chemotherapy and outcome. Berlin-Frankfurt-Münster Relapse Study Group. J Clin Oncol 14 (10): 2812-7, 1996.
- Roy A, Cargill A, Love S, et al.: Outcome after first relapse in childhood acute lymphoblastic leukaemia - lessons from the United Kingdom R2 trial. Br J Haematol 130 (1): 67-75, 2005.
- Rizzari C, Valsecchi MG, Aricò M, et al.: Outcome of very late relapse in children with acute lymphoblastic leukemia. Haematologica 89 (4): 427-34, 2004.
- Nguyen K, Devidas M, Cheng SC, et al.: Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children's Oncology Group study. Leukemia 22 (12): 2142-50, 2008.
- Locatelli F, Schrappe M, Bernardo ME, et al.: How I treat relapsed childhood acute lymphoblastic leukemia. Blood 120 (14): 2807-16, 2012.
- Malempati S, Gaynon PS, Sather H, et al.: Outcome after relapse among children with standard-risk acute lymphoblastic leukemia: Children's Oncology Group study CCG-1952. J Clin Oncol 25 (36): 5800-7, 2007.
- Masurekar AN, Parker CA, Shanyinde M, et al.: Outcome of central nervous system relapses in childhood acute lymphoblastic leukaemia--prospective open cohort analyses of the ALLR3 trial. PLoS One 9 (10): e108107, 2014.
- Parker C, Krishnan S, Hamadeh L, et al.: Outcomes of patients with childhood B-cell precursor acute lymphoblastic leukaemia with late bone marrow relapses: long-term follow-up of the ALLR3 open-label randomised trial. Lancet Haematol 6 (4): e204-e216, 2019.
- Barredo JC, Devidas M, Lauer SJ, et al.: Isolated CNS relapse of acute lymphoblastic leukemia treated with intensive systemic chemotherapy and delayed CNS radiation: a pediatric oncology group study. J Clin Oncol 24 (19): 3142-9, 2006.
- Rubnitz JE, Hijiya N, Zhou Y, et al.: Lack of benefit of early detection of relapse after completion of therapy for acute lymphoblastic leukemia. Pediatr Blood Cancer 44 (2): 138-41, 2005.
- Freyer DR, Devidas M, La M, et al.: Postrelapse survival in childhood acute lymphoblastic leukemia is independent of initial treatment intensity: a report from the Children's Oncology Group. Blood 117 (11): 3010-5, 2011.
- Eckert C, Groeneveld-Krentz S, Kirschner-Schwabe R, et al.: Improving Stratification for Children With Late Bone Marrow B-Cell Acute Lymphoblastic Leukemia Relapses With Refined Response Classification and Integration of Genetics. J Clin Oncol 37 (36): 3493-3506, 2019.
- Meyr F, Escherich G, Mann G, et al.: Outcomes of treatment for relapsed acute lymphoblastic leukaemia in children with Down syndrome. Br J Haematol 162 (1): 98-106, 2013.
- Hitzler JK, He W, Doyle J, et al.: Outcome of transplantation for acute lymphoblastic leukemia in children with Down syndrome. Pediatr Blood Cancer 61 (6): 1126-8, 2014.
- Raetz EA, Borowitz MJ, Devidas M, et al.: Reinduction platform for children with first marrow relapse in acute lymphoblastic lymphoma. J Clin Oncol 26 (24): 3971-8, 2008.
- von Stackelberg A, Völzke E, Kühl JS, et al.: Outcome of children and adolescents with relapsed acute lymphoblastic leukaemia and non-response to salvage protocol therapy: a retrospective analysis of the ALL-REZ BFM Study Group. Eur J Cancer 47 (1): 90-7, 2011.
- Coustan-Smith E, Gajjar A, Hijiya N, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia after first relapse. Leukemia 18 (3): 499-504, 2004.
- Sramkova L, Muzikova K, Fronkova E, et al.: Detectable minimal residual disease before allogeneic hematopoietic stem cell transplantation predicts extremely poor prognosis in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 48 (1): 93-100, 2007.
- Eckert C, von Stackelberg A, Seeger K, et al.: Minimal residual disease after induction is the strongest predictor of prognosis in intermediate risk relapsed acute lymphoblastic leukaemia - long-term results of trial ALL-REZ BFM P95/96. Eur J Cancer 49 (6): 1346-55, 2013.
- Eckert C, Parker C, Moorman AV, et al.: Risk factors and outcomes in children with high-risk B-cell precursor and T-cell relapsed acute lymphoblastic leukaemia: combined analysis of ALLR3 and ALL-REZ BFM 2002 clinical trials. Eur J Cancer 151: 175-189, 2021.
- Paganin M, Zecca M, Fabbri G, et al.: Minimal residual disease is an important predictive factor of outcome in children with relapsed 'high-risk' acute lymphoblastic leukemia. Leukemia 22 (12): 2193-200, 2008.
- Lew G, Chen Y, Lu X, et al.: Outcomes after late bone marrow and very early central nervous system relapse of childhood B-acute lymphoblastic leukemia: a report from the Children's Oncology Group phase III study AALL0433. Haematologica 106 (1): 46-55, 2021.
- Ma X, Edmonson M, Yergeau D, et al.: Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun 6: 6604, 2015.
- Mullighan CG, Zhang J, Kasper LH, et al.: CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471 (7337): 235-9, 2011.
- Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet 54 (9): 1376-1389, 2022.
- Meyer JA, Wang J, Hogan LE, et al.: Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 45 (3): 290-4, 2013.
- Tzoneva G, Perez-Garcia A, Carpenter Z, et al.: Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med 19 (3): 368-71, 2013.
- Hof J, Krentz S, van Schewick C, et al.: Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol 29 (23): 3185-93, 2011.
- Krentz S, Hof J, Mendioroz A, et al.: Prognostic value of genetic alterations in children with first bone marrow relapse of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia 27 (2): 295-304, 2013.
- Irving J, Matheson E, Minto L, et al.: Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 124 (23): 3420-30, 2014.
- Gandemer V, Chevret S, Petit A, et al.: Excellent prognosis of late relapses of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia: lessons from the FRALLE 93 protocol. Haematologica 97 (11): 1743-50, 2012.
- Tallen G, Ratei R, Mann G, et al.: Long-term outcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrug chemotherapy: results of trial ALL-REZ BFM 90. J Clin Oncol 28 (14): 2339-47, 2010.
- Parker C, Waters R, Leighton C, et al.: Effect of mitoxantrone on outcome of children with first relapse of acute lymphoblastic leukaemia (ALL R3): an open-label randomised trial. Lancet 376 (9757): 2009-17, 2010.
- von Stackelberg A, Hartmann R, Bührer C, et al.: High-dose compared with intermediate-dose methotrexate in children with a first relapse of acute lymphoblastic leukemia. Blood 111 (5): 2573-80, 2008.
- Locatelli F, Testi AM, Bernardo ME, et al.: Clofarabine, cyclophosphamide and etoposide as single-course re-induction therapy for children with refractory/multiple relapsed acute lymphoblastic leukaemia. Br J Haematol 147 (3): 371-8, 2009.
- Miano M, Pistorio A, Putti MC, et al.: Clofarabine, cyclophosphamide and etoposide for the treatment of relapsed or resistant acute leukemia in pediatric patients. Leuk Lymphoma 53 (9): 1693-8, 2012.
- Hijiya N, Thomson B, Isakoff MS, et al.: Phase 2 trial of clofarabine in combination with etoposide and cyclophosphamide in pediatric patients with refractory or relapsed acute lymphoblastic leukemia. Blood 118 (23): 6043-9, 2011.
- Bertaina A, Vinti L, Strocchio L, et al.: The combination of bortezomib with chemotherapy to treat relapsed/refractory acute lymphoblastic leukaemia of childhood. Br J Haematol 176 (4): 629-636, 2017.
- Messinger YH, Gaynon PS, Sposto R, et al.: Bortezomib with chemotherapy is highly active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Blood 120 (2): 285-90, 2012.
- Horton TM, Whitlock JA, Lu X, et al.: Bortezomib reinduction chemotherapy in high-risk ALL in first relapse: a report from the Children's Oncology Group. Br J Haematol 186 (2): 274-285, 2019.
- Berg SL, Blaney SM, Devidas M, et al.: Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children's Oncology Group. J Clin Oncol 23 (15): 3376-82, 2005.
- Zwaan CM, Kowalczyk J, Schmitt C, et al.: Safety and efficacy of nelarabine in children and young adults with relapsed or refractory T-lineage acute lymphoblastic leukaemia or T-lineage lymphoblastic lymphoma: results of a phase 4 study. Br J Haematol 179 (2): 284-293, 2017.
- Commander LA, Seif AE, Insogna IG, et al.: Salvage therapy with nelarabine, etoposide, and cyclophosphamide in relapsed/refractory paediatric T-cell lymphoblastic leukaemia and lymphoma. Br J Haematol 150 (3): 345-51, 2010.
- Whitlock JA, Malvar J, Dalla-Pozza L, et al.: Nelarabine, etoposide, and cyclophosphamide in relapsed pediatric T-acute lymphoblastic leukemia and T-lymphoblastic lymphoma (study T2008-002 NECTAR). Pediatr Blood Cancer 69 (11): e29901, 2022.
- Sun W, Malvar J, Sposto R, et al.: Outcome of children with multiply relapsed B-cell acute lymphoblastic leukemia: a therapeutic advances in childhood leukemia & lymphoma study. Leukemia 32 (11): 2316-2325, 2018.
- Eapen M, Raetz E, Zhang MJ, et al.: Outcomes after HLA-matched sibling transplantation or chemotherapy in children with B-precursor acute lymphoblastic leukemia in a second remission: a collaborative study of the Children's Oncology Group and the Center for International Blood and Marrow Transplant Research. Blood 107 (12): 4961-7, 2006.
- Barrett AJ, Horowitz MM, Pollock BH, et al.: Bone marrow transplants from HLA-identical siblings as compared with chemotherapy for children with acute lymphoblastic leukemia in a second remission. N Engl J Med 331 (19): 1253-8, 1994.
- Uderzo C, Valsecchi MG, Bacigalupo A, et al.: Treatment of childhood acute lymphoblastic leukemia in second remission with allogeneic bone marrow transplantation and chemotherapy: ten-year experience of the Italian Bone Marrow Transplantation Group and the Italian Pediatric Hematology Oncology Association. J Clin Oncol 13 (2): 352-8, 1995.
- Harrison G, Richards S, Lawson S, et al.: Comparison of allogeneic transplant versus chemotherapy for relapsed childhood acute lymphoblastic leukaemia in the MRC UKALL R1 trial. MRC Childhood Leukaemia Working Party. Ann Oncol 11 (8): 999-1006, 2000.
- Bunin N, Carston M, Wall D, et al.: Unrelated marrow transplantation for children with acute lymphoblastic leukemia in second remission. Blood 99 (9): 3151-7, 2002.
- Borgmann A, von Stackelberg A, Hartmann R, et al.: Unrelated donor stem cell transplantation compared with chemotherapy for children with acute lymphoblastic leukemia in a second remission: a matched-pair analysis. Blood 101 (10): 3835-9, 2003.
- Saarinen-Pihkala UM, Heilmann C, Winiarski J, et al.: Pathways through relapses and deaths of children with acute lymphoblastic leukemia: role of allogeneic stem-cell transplantation in Nordic data. J Clin Oncol 24 (36): 5750-62, 2006.
- Thomson B, Park JR, Felgenhauer J, et al.: Toxicity and efficacy of intensive chemotherapy for children with acute lymphoblastic leukemia (ALL) after first bone marrow or extramedullary relapse. Pediatr Blood Cancer 43 (5): 571-9, 2004.
- Hahn T, Wall D, Camitta B, et al.: The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute lymphoblastic leukemia in children: an evidence-based review. Biol Blood Marrow Transplant 11 (11): 823-61, 2005.
- Locatelli F, Zugmaier G, Rizzari C, et al.: Effect of Blinatumomab vs Chemotherapy on Event-Free Survival Among Children With High-risk First-Relapse B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 325 (9): 843-854, 2021.
- Brown PA, Ji L, Xu X, et al.: Effect of Postreinduction Therapy Consolidation With Blinatumomab vs Chemotherapy on Disease-Free Survival in Children, Adolescents, and Young Adults With First Relapse of B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 325 (9): 833-842, 2021.
- Borgmann A, Baumgarten E, Schmid H, et al.: Allogeneic bone marrow transplantation for a subset of children with acute lymphoblastic leukemia in third remission: a conceivable alternative? Bone Marrow Transplant 20 (11): 939-44, 1997.
- Schroeder H, Gustafsson G, Saarinen-Pihkala UM, et al.: Allogeneic bone marrow transplantation in second remission of childhood acute lymphoblastic leukemia: a population-based case control study from the Nordic countries. Bone Marrow Transplant 23 (6): 555-60, 1999.
- van den Berg H, de Groot-Kruseman HA, Damen-Korbijn CM, et al.: Outcome after first relapse in children with acute lymphoblastic leukemia: a report based on the Dutch Childhood Oncology Group (DCOG) relapse all 98 protocol. Pediatr Blood Cancer 57 (2): 210-6, 2011.
- Beck JC, Cao Q, Trotz B, et al.: Allogeneic hematopoietic cell transplantation outcomes for children with B-precursor acute lymphoblastic leukemia and early or late BM relapse. Bone Marrow Transplant 46 (7): 950-5, 2011.
- Eckert C, Henze G, Seeger K, et al.: Use of allogeneic hematopoietic stem-cell transplantation based on minimal residual disease response improves outcomes for children with relapsed acute lymphoblastic leukemia in the intermediate-risk group. J Clin Oncol 31 (21): 2736-42, 2013.
- Hogan LE, Brown PA, Ji L, et al.: Children's Oncology Group AALL1331: Phase III Trial of Blinatumomab in Children, Adolescents, and Young Adults With Low-Risk B-Cell ALL in First Relapse. J Clin Oncol 41 (25): 4118-4129, 2023.
- Aubert L, Petit A, Bertrand Y, et al.: Therapeutic approach and outcome of children with Philadelphia chromosome-positive acute lymphoblastic leukemia at first relapse in the era of tyrosine kinase inhibitors: An SFCE retrospective study. Pediatr Blood Cancer 69 (2): e29441, 2022.
- Burke MJ, Verneris MR, Le Rademacher J, et al.: Transplant Outcomes for Children with T Cell Acute Lymphoblastic Leukemia in Second Remission: A Report from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant 21 (12): 2154-9, 2015.
- Gaynon PS: Childhood acute lymphoblastic leukaemia and relapse. Br J Haematol 131 (5): 579-87, 2005.
- Jeha S, Goto H, Baruchel A, et al.: Patient-Level Meta-analysis of Clofarabine in Acute Lymphoblastic Leukemia. Adv Ther 40 (12): 5447-5463, 2023.
- Pullarkat VA, Lacayo NJ, Jabbour E, et al.: Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancer Discov 11 (6): 1440-1453, 2021.
- Woolfrey AE, Anasetti C, Storer B, et al.: Factors associated with outcome after unrelated marrow transplantation for treatment of acute lymphoblastic leukemia in children. Blood 99 (6): 2002-8, 2002.
- Afify Z, Hunt L, Green A, et al.: Factors affecting the outcome of stem cell transplantation from unrelated donors for childhood acute lymphoblastic leukemia in third remission. Bone Marrow Transplant 35 (11): 1041-7, 2005.
- Gassas A, Ishaqi MK, Afzal S, et al.: Outcome of haematopoietic stem cell transplantation for paediatric acute lymphoblastic leukaemia in third complete remission: a vital role for graft-versus-host-disease/ graft-versus-leukaemia effect in survival. Br J Haematol 140 (1): 86-9, 2008.
- Nemecek ER, Ellis K, He W, et al.: Outcome of myeloablative conditioning and unrelated donor hematopoietic cell transplantation for childhood acute lymphoblastic leukemia in third remission. Biol Blood Marrow Transplant 17 (12): 1833-40, 2011.
- Kato M, Horikoshi Y, Okamoto Y, et al.: Second allogeneic hematopoietic SCT for relapsed ALL in children. Bone Marrow Transplant 47 (10): 1307-11, 2012.
- Bhojwani D, Sposto R, Shah NN, et al.: Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. Leukemia 33 (4): 884-892, 2019.
- Pulsipher MA: Are CAR T cells better than antibody or HCT therapy in B-ALL? Hematology Am Soc Hematol Educ Program 2018 (1): 16-24, 2018.
- Contreras CF, Higham CS, Behnert A, et al.: Clinical utilization of blinatumomab and inotuzumab immunotherapy in children with relapsed or refractory B-acute lymphoblastic leukemia. Pediatr Blood Cancer 68 (1): e28718, 2021.
- Locatelli F, Maschan A, Boissel N, et al.: Pediatric patients with acute lymphoblastic leukemia treated with blinatumomab in a real-world setting: Results from the NEUF study. Pediatr Blood Cancer 69 (4): e29562, 2022.
- O'Brien MM, Ji L, Shah NN, et al.: Phase II Trial of Inotuzumab Ozogamicin in Children and Adolescents With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia: Children's Oncology Group Protocol AALL1621. J Clin Oncol 40 (9): 956-967, 2022.
- Oliansky DM, Camitta B, Gaynon P, et al.: Role of cytotoxic therapy with hematopoietic stem cell transplantation in the treatment of pediatric acute lymphoblastic leukemia: update of the 2005 evidence-based review. Biol Blood Marrow Transplant 18 (4): 505-22, 2012.
- Qayed M, Ahn KW, Kitko CL, et al.: A validated pediatric disease risk index for allogeneic hematopoietic cell transplantation. Blood 137 (7): 983-993, 2021.
- Davies SM, Ramsay NK, Klein JP, et al.: Comparison of preparative regimens in transplants for children with acute lymphoblastic leukemia. J Clin Oncol 18 (2): 340-7, 2000.
- Bunin N, Aplenc R, Kamani N, et al.: Randomized trial of busulfan vs total body irradiation containing conditioning regimens for children with acute lymphoblastic leukemia: a Pediatric Blood and Marrow Transplant Consortium study. Bone Marrow Transplant 32 (6): 543-8, 2003.
- Bader P, Salzmann-Manrique E, Balduzzi A, et al.: More precisely defining risk peri-HCT in pediatric ALL: pre- vs post-MRD measures, serial positivity, and risk modeling. Blood Adv 3 (21): 3393-3405, 2019.
- Peters C, Dalle JH, Locatelli F, et al.: Total Body Irradiation or Chemotherapy Conditioning in Childhood ALL: A Multinational, Randomized, Noninferiority Phase III Study. J Clin Oncol 39 (4): 295-307, 2021.
- Gassas A, Sung L, Saunders EF, et al.: Comparative outcome of hematopoietic stem cell transplantation for pediatric acute lymphoblastic leukemia following cyclophosphamide and total body irradiation or VP16 and total body irradiation conditioning regimens. Bone Marrow Transplant 38 (11): 739-43, 2006.
- Tracey J, Zhang MJ, Thiel E, et al.: Transplantation conditioning regimens and outcomes after allogeneic hematopoietic cell transplantation in children and adolescents with acute lymphoblastic leukemia. Biol Blood Marrow Transplant 19 (2): 255-9, 2013.
- Bakr M, Rasheed W, Mohamed SY, et al.: Allogeneic hematopoietic stem cell transplantation in adolescent and adult patients with high-risk T cell acute lymphoblastic leukemia. Biol Blood Marrow Transplant 18 (12): 1897-904, 2012.
- Marks DI, Forman SJ, Blume KG, et al.: A comparison of cyclophosphamide and total body irradiation with etoposide and total body irradiation as conditioning regimens for patients undergoing sibling allografting for acute lymphoblastic leukemia in first or second complete remission. Biol Blood Marrow Transplant 12 (4): 438-53, 2006.
- Esiashvili N, Lu X, Ulin K, et al.: Higher Reported Lung Dose Received During Total Body Irradiation for Allogeneic Hematopoietic Stem Cell Transplantation in Children With Acute Lymphoblastic Leukemia Is Associated With Inferior Survival: A Report from the Children's Oncology Group. Int J Radiat Oncol Biol Phys 104 (3): 513-521, 2019.
- Duval M, Klein JP, He W, et al.: Hematopoietic stem-cell transplantation for acute leukemia in relapse or primary induction failure. J Clin Oncol 28 (23): 3730-8, 2010.
- Shen Z, Gu X, Mao W, et al.: Influence of pre-transplant minimal residual disease on prognosis after Allo-SCT for patients with acute lymphoblastic leukemia: systematic review and meta-analysis. BMC Cancer 18 (1): 755, 2018.
- Balduzzi A, Di Maio L, Silvestri D, et al.: Minimal residual disease before and after transplantation for childhood acute lymphoblastic leukaemia: is there any room for intervention? Br J Haematol 164 (3): 396-408, 2014.
- Goulden N, Bader P, Van Der Velden V, et al.: Minimal residual disease prior to stem cell transplant for childhood acute lymphoblastic leukaemia. Br J Haematol 122 (1): 24-9, 2003.
- Bader P, Kreyenberg H, Henze GH, et al.: Prognostic value of minimal residual disease quantification before allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia: the ALL-REZ BFM Study Group. J Clin Oncol 27 (3): 377-84, 2009.
- Leung W, Pui CH, Coustan-Smith E, et al.: Detectable minimal residual disease before hematopoietic cell transplantation is prognostic but does not preclude cure for children with very-high-risk leukemia. Blood 120 (2): 468-72, 2012.
- Ruggeri A, Michel G, Dalle JH, et al.: Impact of pretransplant minimal residual disease after cord blood transplantation for childhood acute lymphoblastic leukemia in remission: an Eurocord, PDWP-EBMT analysis. Leukemia 26 (12): 2455-61, 2012.
- Bachanova V, Burke MJ, Yohe S, et al.: Unrelated cord blood transplantation in adult and pediatric acute lymphoblastic leukemia: effect of minimal residual disease on relapse and survival. Biol Blood Marrow Transplant 18 (6): 963-8, 2012.
- Sutton R, Shaw PJ, Venn NC, et al.: Persistent MRD before and after allogeneic BMT predicts relapse in children with acute lymphoblastic leukaemia. Br J Haematol 168 (3): 395-404, 2015.
- Sanchez-Garcia J, Serrano J, Serrano-Lopez J, et al.: Quantification of minimal residual disease levels by flow cytometry at time of transplant predicts outcome after myeloablative allogeneic transplantation in ALL. Bone Marrow Transplant 48 (3): 396-402, 2013.
- Ifversen M, Turkiewicz D, Marquart HV, et al.: Low burden of minimal residual disease prior to transplantation in children with very high risk acute lymphoblastic leukaemia: The NOPHO ALL2008 experience. Br J Haematol 184 (6): 982-993, 2019.
- Bader P, Kreyenberg H, von Stackelberg A, et al.: Monitoring of minimal residual disease after allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia allows for the identification of impending relapse: results of the ALL-BFM-SCT 2003 trial. J Clin Oncol 33 (11): 1275-84, 2015.
- Pulsipher MA, Langholz B, Wall DA, et al.: Risk factors and timing of relapse after allogeneic transplantation in pediatric ALL: for whom and when should interventions be tested? Bone Marrow Transplant 50 (9): 1173-9, 2015.
- Pulsipher MA, Carlson C, Langholz B, et al.: IgH-V(D)J NGS-MRD measurement pre- and early post-allotransplant defines very low- and very high-risk ALL patients. Blood 125 (22): 3501-8, 2015.
- Liu J, Wang Y, Xu LP, et al.: Monitoring mixed lineage leukemia expression may help identify patients with mixed lineage leukemia--rearranged acute leukemia who are at high risk of relapse after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 20 (7): 929-36, 2014.
- Locatelli F, Zecca M, Messina C, et al.: Improvement over time in outcome for children with acute lymphoblastic leukemia in second remission given hematopoietic stem cell transplantation from unrelated donors. Leukemia 16 (11): 2228-37, 2002.
- Saarinen-Pihkala UM, Gustafsson G, Ringdén O, et al.: No disadvantage in outcome of using matched unrelated donors as compared with matched sibling donors for bone marrow transplantation in children with acute lymphoblastic leukemia in second remission. J Clin Oncol 19 (14): 3406-14, 2001.
- Muñoz A, Diaz-Heredia C, Diaz MA, et al.: Allogeneic hemopoietic stem cell transplantation for childhood acute lymphoblastic leukemia in second complete remission-similar outcomes after matched related and unrelated donor transplant: a study of the Spanish Working Party for Blood and Marrow Transplantation in Children (Getmon). Pediatr Hematol Oncol 25 (4): 245-59, 2008.
- Jacobsohn DA, Hewlett B, Ranalli M, et al.: Outcomes of unrelated cord blood transplants and allogeneic-related hematopoietic stem cell transplants in children with high-risk acute lymphocytic leukemia. Bone Marrow Transplant 34 (10): 901-7, 2004.
- Kurtzberg J, Prasad VK, Carter SL, et al.: Results of the Cord Blood Transplantation Study (COBLT): clinical outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with hematologic malignancies. Blood 112 (10): 4318-27, 2008.
- Peters C, Schrappe M, von Stackelberg A, et al.: Stem-cell transplantation in children with acute lymphoblastic leukemia: A prospective international multicenter trial comparing sibling donors with matched unrelated donors-The ALL-SCT-BFM-2003 trial. J Clin Oncol 33 (11): 1265-74, 2015.
- Smith AR, Baker KS, Defor TE, et al.: Hematopoietic cell transplantation for children with acute lymphoblastic leukemia in second complete remission: similar outcomes in recipients of unrelated marrow and umbilical cord blood versus marrow from HLA matched sibling donors. Biol Blood Marrow Transplant 15 (9): 1086-93, 2009.
- Zhang MJ, Davies SM, Camitta BM, et al.: Comparison of outcomes after HLA-matched sibling and unrelated donor transplantation for children with high-risk acute lymphoblastic leukemia. Biol Blood Marrow Transplant 18 (8): 1204-10, 2012.
- Gassas A, Sung L, Saunders EF, et al.: Graft-versus-leukemia effect in hematopoietic stem cell transplantation for pediatric acute lymphoblastic leukemia: significantly lower relapse rate in unrelated transplantations. Bone Marrow Transplant 40 (10): 951-5, 2007.
- Harvey J, Green A, Cornish J, et al.: Improved survival in matched unrelated donor transplant for childhood ALL since the introduction of high-resolution matching at HLA class I and II. Bone Marrow Transplant 47 (10): 1294-300, 2012.
- Majhail NS, Chitphakdithai P, Logan B, et al.: Significant improvement in survival after unrelated donor hematopoietic cell transplantation in the recent era. Biol Blood Marrow Transplant 21 (1): 142-50, 2015.
- MacMillan ML, Davies SM, Nelson GO, et al.: Twenty years of unrelated donor bone marrow transplantation for pediatric acute leukemia facilitated by the National Marrow Donor Program. Biol Blood Marrow Transplant 14 (9 Suppl): 16-22, 2008.
- Davies SM, Wang D, Wang T, et al.: Recent decrease in acute graft-versus-host disease in children with leukemia receiving unrelated donor bone marrow transplants. Biol Blood Marrow Transplant 15 (3): 360-6, 2009.
- Eapen M, Rubinstein P, Zhang MJ, et al.: Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: a comparison study. Lancet 369 (9577): 1947-54, 2007.
- Klingebiel T, Handgretinger R, Lang P, et al.: Haploidentical transplantation for acute lymphoblastic leukemia in childhood. Blood Rev 18 (3): 181-92, 2004.
- Ruggeri A, Galimard JE, Paina O, et al.: Outcomes of Unmanipulated Haploidentical Transplantation Using Post-Transplant Cyclophosphamide (PT-Cy) in Pediatric Patients With Acute Lymphoblastic Leukemia. Transplant Cell Ther 27 (5): 424.e1-424.e9, 2021.
- Locatelli F, Merli P, Pagliara D, et al.: Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood 130 (5): 677-685, 2017.
- Bertaina A, Zecca M, Buldini B, et al.: Unrelated donor vs HLA-haploidentical α/β T-cell- and B-cell-depleted HSCT in children with acute leukemia. Blood 132 (24): 2594-2607, 2018.
- Pulsipher MA, Ahn KW, Bunin NJ, et al.: KIR-favorable TCR-αβ/CD19-depleted haploidentical HCT in children with ALL/AML/MDS: primary analysis of the PTCTC ONC1401 trial. Blood 140 (24): 2556-2572, 2022.
- Gustafsson Jernberg A, Remberger M, Ringdén O, et al.: Graft-versus-leukaemia effect in children: chronic GVHD has a significant impact on relapse and survival. Bone Marrow Transplant 31 (3): 175-81, 2003.
- Dini G, Zecca M, Balduzzi A, et al.: No difference in outcome between children and adolescents transplanted for acute lymphoblastic leukemia in second remission. Blood 118 (25): 6683-90, 2011.
- Pulsipher MA, Langholz B, Wall DA, et al.: The addition of sirolimus to tacrolimus/methotrexate GVHD prophylaxis in children with ALL: a phase 3 Children's Oncology Group/Pediatric Blood and Marrow Transplant Consortium trial. Blood 123 (13): 2017-25, 2014.
- Yeshurun M, Weisdorf D, Rowe JM, et al.: The impact of the graft-versus-leukemia effect on survival in acute lymphoblastic leukemia. Blood Adv 3 (4): 670-680, 2019.
- Pulsipher MA, Bader P, Klingebiel T, et al.: Allogeneic transplantation for pediatric acute lymphoblastic leukemia: the emerging role of peritransplantation minimal residual disease/chimerism monitoring and novel chemotherapeutic, molecular, and immune approaches aimed at preventing relapse. Biol Blood Marrow Transplant 15 (1 Suppl): 62-71, 2008.
- Lankester AC, Bierings MB, van Wering ER, et al.: Preemptive alloimmune intervention in high-risk pediatric acute lymphoblastic leukemia patients guided by minimal residual disease level before stem cell transplantation. Leukemia 24 (8): 1462-9, 2010.
- Horn B, Soni S, Khan S, et al.: Feasibility study of preemptive withdrawal of immunosuppression based on chimerism testing in children undergoing myeloablative allogeneic transplantation for hematologic malignancies. Bone Marrow Transplant 43 (6): 469-76, 2009.
- Pochon C, Oger E, Michel G, et al.: Follow-up of post-transplant minimal residual disease and chimerism in childhood lymphoblastic leukaemia: 90 d to react. Br J Haematol 169 (2): 249-61, 2015.
- Bader P, Kreyenberg H, Hoelle W, et al.: Increasing mixed chimerism is an important prognostic factor for unfavorable outcome in children with acute lymphoblastic leukemia after allogeneic stem-cell transplantation: possible role for pre-emptive immunotherapy? J Clin Oncol 22 (9): 1696-705, 2004.
- Gandemer V, Pochon C, Oger E, et al.: Clinical value of pre-transplant minimal residual disease in childhood lymphoblastic leukaemia: the results of the French minimal residual disease-guided protocol. Br J Haematol 165 (3): 392-401, 2014.
- Rubin J, Vettenranta K, Vettenranta J, et al.: Use of intrathecal chemoprophylaxis in children after SCT and the risk of central nervous system relapse. Bone Marrow Transplant 46 (3): 372-8, 2011.
- Thompson CB, Sanders JE, Flournoy N, et al.: The risks of central nervous system relapse and leukoencephalopathy in patients receiving marrow transplants for acute leukemia. Blood 67 (1): 195-9, 1986.
- Oshima K, Kanda Y, Yamashita T, et al.: Central nervous system relapse of leukemia after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 14 (10): 1100-7, 2008.
- Ruutu T, Corradini P, Gratwohl A, et al.: Use of intrathecal prophylaxis in allogeneic haematopoietic stem cell transplantation for malignant blood diseases: a survey of the European Group for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant 35 (2): 121-4, 2005.
- Maude SL, Laetsch TW, Buechner J, et al.: Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 378 (5): 439-448, 2018.
- Mehta J, Powles R, Treleaven J, et al.: Outcome of acute leukemia relapsing after bone marrow transplantation: utility of second transplants and adoptive immunotherapy. Bone Marrow Transplant 19 (7): 709-19, 1997.
- Kuhlen M, Willasch AM, Dalle JH, et al.: Outcome of relapse after allogeneic HSCT in children with ALL enrolled in the ALL-SCT 2003/2007 trial. Br J Haematol 180 (1): 82-89, 2018.
- Eapen M, Giralt SA, Horowitz MM, et al.: Second transplant for acute and chronic leukemia relapsing after first HLA-identical sibling transplant. Bone Marrow Transplant 34 (8): 721-7, 2004.
- Bosi A, Laszlo D, Labopin M, et al.: Second allogeneic bone marrow transplantation in acute leukemia: results of a survey by the European Cooperative Group for Blood and Marrow Transplantation. J Clin Oncol 19 (16): 3675-84, 2001.
- Willasch AM, Salzmann-Manrique E, Krenn T, et al.: Treatment of relapse after allogeneic stem cell transplantation in children and adolescents with ALL: the Frankfurt experience. Bone Marrow Transplant 52 (2): 201-208, 2017.
- Yaniv I, Krauss AC, Beohou E, et al.: Second Hematopoietic Stem Cell Transplantation for Post-Transplantation Relapsed Acute Leukemia in Children: A Retrospective EBMT-PDWP Study. Biol Blood Marrow Transplant 24 (8): 1629-1642, 2018.
- Nishikawa T, Inagaki J, Nagatoshi Y, et al.: The second therapeutic trial for children with hematological malignancies who relapsed after their first allogeneic SCT: long-term outcomes. Pediatr Transplant 16 (7): 722-8, 2012.
- Bajwa R, Schechter T, Soni S, et al.: Outcome of children who experience disease relapse following allogeneic hematopoietic SCT for hematologic malignancies. Bone Marrow Transplant 48 (5): 661-5, 2013.
- Schechter T, Avila L, Frangoul H, et al.: Effect of acute graft-versus-host disease on the outcome of second allogeneic hematopoietic stem cell transplant in children. Leuk Lymphoma 54 (1): 105-9, 2013.
- Pulsipher MA, Boucher KM, Wall D, et al.: Reduced-intensity allogeneic transplantation in pediatric patients ineligible for myeloablative therapy: results of the Pediatric Blood and Marrow Transplant Consortium Study ONC0313. Blood 114 (7): 1429-36, 2009.
- Collins RH, Goldstein S, Giralt S, et al.: Donor leukocyte infusions in acute lymphocytic leukemia. Bone Marrow Transplant 26 (5): 511-6, 2000.
- Levine JE, Barrett AJ, Zhang MJ, et al.: Donor leukocyte infusions to treat hematologic malignancy relapse following allo-SCT in a pediatric population. Bone Marrow Transplant 42 (3): 201-5, 2008.
- Bhadri VA, McGregor MR, Venn NC, et al.: Isolated testicular relapse after allo-SCT in boys with ALL: outcome without second transplant. Bone Marrow Transplant 45 (2): 397-9, 2010.
- von Stackelberg A, Locatelli F, Zugmaier G, et al.: Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. J Clin Oncol 34 (36): 4381-4389, 2016.
- Queudeville M, Stein AS, Locatelli F, et al.: Low leukemia burden improves blinatumomab efficacy in patients with relapsed/refractory B-cell acute lymphoblastic leukemia. Cancer 129 (9): 1384-1393, 2023.
- Kantarjian H, Thomas D, Jorgensen J, et al.: Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer 119 (15): 2728-36, 2013.
- Kantarjian HM, DeAngelo DJ, Stelljes M, et al.: Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N Engl J Med 375 (8): 740-53, 2016.
- Pennesi E, Michels N, Brivio E, et al.: Inotuzumab ozogamicin as single agent in pediatric patients with relapsed and refractory acute lymphoblastic leukemia: results from a phase II trial. Leukemia 36 (6): 1516-1524, 2022.
- Kebriaei P, Cutler C, de Lima M, et al.: Management of important adverse events associated with inotuzumab ozogamicin: expert panel review. Bone Marrow Transplant 53 (4): 449-456, 2018.
- Maury S, Chevret S, Thomas X, et al.: Rituximab in B-Lineage Adult Acute Lymphoblastic Leukemia. N Engl J Med 375 (11): 1044-53, 2016.
- Sasaki K, Kantarjian HM, Morita K, et al.: Hyper-CVAD plus ofatumumab versus hyper-CVAD plus rituximab as frontline therapy in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: A propensity score analysis. Cancer 127 (18): 3381-3389, 2021.
- Grupp SA, Kalos M, Barrett D, et al.: Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368 (16): 1509-18, 2013.
- Maude SL, Frey N, Shaw PA, et al.: Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371 (16): 1507-17, 2014.
- Fitzgerald JC, Weiss SL, Maude SL, et al.: Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit Care Med 45 (2): e124-e131, 2017.
- Kadauke S, Myers RM, Li Y, et al.: Risk-Adapted Preemptive Tocilizumab to Prevent Severe Cytokine Release Syndrome After CTL019 for Pediatric B-Cell Acute Lymphoblastic Leukemia: A Prospective Clinical Trial. J Clin Oncol 39 (8): 920-930, 2021.
- Shalabi H, Wolters PL, Martin S, et al.: Systematic Evaluation of Neurotoxicity in Children and Young Adults Undergoing CD22 Chimeric Antigen Receptor T-Cell Therapy. J Immunother 41 (7): 350-358, 2018.
- Gardner RA, Finney O, Annesley C, et al.: Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 129 (25): 3322-3331, 2017.
- Levine JE, Grupp SA, Pulsipher MA, et al.: Pooled safety analysis of tisagenlecleucel in children and young adults with B cell acute lymphoblastic leukemia. J Immunother Cancer 9 (8): , 2021.
- McNerney KO, Si Lim SJ, Ishikawa K, et al.: HLH-like toxicities predict poor survival after the use of tisagenlecleucel in children and young adults with B-ALL. Blood Adv 7 (12): 2758-2771, 2023.
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al.: T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385 (9967): 517-28, 2015.
- V Stackelberg A, Jäschke K, Jousseaume E, et al.: Tisagenlecleucel vs. historical standard of care in children and young adult patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia. Leukemia 37 (12): 2346-2355, 2023.
- Pasquini MC, Hu ZH, Curran K, et al.: Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv 4 (21): 5414-5424, 2020.
- Ghorashian S, Jacoby E, De Moerloose B, et al.: Tisagenlecleucel therapy for relapsed or refractory B-cell acute lymphoblastic leukaemia in infants and children younger than 3 years of age at screening: an international, multicentre, retrospective cohort study. Lancet Haematol 9 (10): e766-e775, 2022.
- Laetsch TW, Maude SL, Rives S, et al.: Three-Year Update of Tisagenlecleucel in Pediatric and Young Adult Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia in the ELIANA Trial. J Clin Oncol 41 (9): 1664-1669, 2023.
- Shah NN, Lee DW, Yates B, et al.: Long-Term Follow-Up of CD19-CAR T-Cell Therapy in Children and Young Adults With B-ALL. J Clin Oncol 39 (15): 1650-1659, 2021.
- Curran KJ, Margossian SP, Kernan NA, et al.: Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood 134 (26): 2361-2368, 2019.
- Myers RM, Li Y, Barz Leahy A, et al.: Humanized CD19-Targeted Chimeric Antigen Receptor (CAR) T Cells in CAR-Naive and CAR-Exposed Children and Young Adults With Relapsed or Refractory Acute Lymphoblastic Leukemia. J Clin Oncol 39 (27): 3044-3055, 2021.
- Leahy AB, Newman H, Li Y, et al.: CD19-targeted chimeric antigen receptor T-cell therapy for CNS relapsed or refractory acute lymphocytic leukaemia: a post-hoc analysis of pooled data from five clinical trials. Lancet Haematol 8 (10): e711-e722, 2021.
- Fabrizio VA, Phillips CL, Lane A, et al.: Tisagenlecleucel outcomes in relapsed/refractory extramedullary ALL: a Pediatric Real World CAR Consortium Report. Blood Adv 6 (2): 600-610, 2022.
- Jacoby E, Ghorashian S, Vormoor B, et al.: CD19 CAR T-cells for pediatric relapsed acute lymphoblastic leukemia with active CNS involvement: a retrospective international study. Leukemia 36 (6): 1525-1532, 2022.
- Myers RM, Taraseviciute A, Steinberg SM, et al.: Blinatumomab Nonresponse and High-Disease Burden Are Associated With Inferior Outcomes After CD19-CAR for B-ALL. J Clin Oncol 40 (9): 932-944, 2022.
- Holland EM, Yates B, Ling A, et al.: Characterization of extramedullary disease in B-ALL and response to CAR T-cell therapy. Blood Adv 6 (7): 2167-2182, 2022.
- Leahy AB, Devine KJ, Li Y, et al.: Impact of high-risk cytogenetics on outcomes for children and young adults receiving CD19-directed CAR T-cell therapy. Blood 139 (14): 2173-2185, 2022.
- Schultz LM, Baggott C, Prabhu S, et al.: Disease Burden Affects Outcomes in Pediatric and Young Adult B-Cell Lymphoblastic Leukemia After Commercial Tisagenlecleucel: A Pediatric Real-World Chimeric Antigen Receptor Consortium Report. J Clin Oncol 40 (9): 945-955, 2022.
- Pulsipher MA, Han X, Maude SL, et al.: Next-Generation Sequencing of Minimal Residual Disease for Predicting Relapse after Tisagenlecleucel in Children and Young Adults with Acute Lymphoblastic Leukemia. Blood Cancer Discov 3 (1): 66-81, 2022.
- Stefanski HE, Eaton A, Baggott C, et al.: Higher doses of tisagenlecleucel are associated with improved outcomes: a report from the pediatric real-world CAR consortium. Blood Adv 7 (4): 541-548, 2023.
- Bader P, Rossig C, Hutter M, et al.: CD19 CAR T cells are an effective therapy for posttransplant relapse in patients with B-lineage ALL: real-world data from Germany. Blood Adv 7 (11): 2436-2448, 2023.
- Lamble AJ, Myers RM, Taraseviciute A, et al.: Preinfusion factors impacting relapse immunophenotype following CD19 CAR T cells. Blood Adv 7 (4): 575-585, 2023.
- Schultz LM, Eaton A, Baggott C, et al.: Outcomes After Nonresponse and Relapse Post-Tisagenlecleucel in Children, Adolescents, and Young Adults With B-Cell Acute Lymphoblastic Leukemia. J Clin Oncol 41 (2): 354-363, 2023.
- Del Bufalo F, Becilli M, Rosignoli C, et al.: Allogeneic, donor-derived, second-generation, CD19-directed CAR-T cells for the treatment of pediatric relapsed/refractory BCP-ALL. Blood 142 (2): 146-157, 2023.
- Summers C, Wu QV, Annesley C, et al.: Hematopoietic Cell Transplantation after CD19 Chimeric Antigen Receptor T Cell-Induced Acute Lymphoblastic Lymphoma Remission Confers a Leukemia-Free Survival Advantage. Transplant Cell Ther 28 (1): 21-29, 2022.
- Zhang Y, Chen H, Song Y, et al.: Chimeric antigens receptor T cell therapy as a bridge to haematopoietic stem cell transplantation for refractory/ relapsed B-cell acute lymphomalastic leukemia. Br J Haematol 189 (1): 146-152, 2020.
- Sotillo E, Barrett DM, Black KL, et al.: Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov 5 (12): 1282-95, 2015.
- Fry TJ, Shah NN, Orentas RJ, et al.: CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med 24 (1): 20-28, 2018.
- Pan J, Niu Q, Deng B, et al.: CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia 33 (12): 2854-2866, 2019.
- Hu GH, Zhao XY, Zuo YX, et al.: Unmanipulated haploidentical hematopoietic stem cell transplantation is an excellent option for children and young adult relapsed/refractory Philadelphia chromosome-negative B-cell acute lymphoblastic leukemia after CAR-T-cell therapy. Leukemia 35 (11): 3092-3100, 2021.
- Shah NN, Highfill SL, Shalabi H, et al.: CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J Clin Oncol 38 (17): 1938-1950, 2020.
- Wang T, Tang Y, Cai J, et al.: Coadministration of CD19- and CD22-Directed Chimeric Antigen Receptor T-Cell Therapy in Childhood B-Cell Acute Lymphoblastic Leukemia: A Single-Arm, Multicenter, Phase II Trial. J Clin Oncol 41 (9): 1670-1683, 2023.
- Pan J, Tang K, Luo Y, et al.: Sequential CD19 and CD22 chimeric antigen receptor T-cell therapy for childhood refractory or relapsed B-cell acute lymphocytic leukaemia: a single-arm, phase 2 study. Lancet Oncol 24 (11): 1229-1241, 2023.
- Spiegel JY, Patel S, Muffly L, et al.: CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat Med 27 (8): 1419-1431, 2021.
- Cordoba S, Onuoha S, Thomas S, et al.: CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: a phase 1 trial. Nat Med 27 (10): 1797-1805, 2021.
- Shalabi H, Qin H, Su A, et al.: CD19/22 CAR T cells in children and young adults with B-ALL: phase 1 results and development of a novel bicistronic CAR. Blood 140 (5): 451-463, 2022.
- Möricke A, Zimmermann M, Reiter A, et al.: Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 24 (2): 265-84, 2010.
- Silverman LB, Stevenson KE, O'Brien JE, et al.: Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia 24 (2): 320-34, 2010.
- Pui CH, Pei D, Sandlund JT, et al.: Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 24 (2): 371-82, 2010.
- Ritchey AK, Pollock BH, Lauer SJ, et al.: Improved survival of children with isolated CNS relapse of acute lymphoblastic leukemia: a pediatric oncology group study . J Clin Oncol 17 (12): 3745-52, 1999.
- Domenech C, Mercier M, Plouvier E, et al.: First isolated extramedullary relapse in children with B-cell precursor acute lymphoblastic leukaemia: results of the Cooprall-97 study. Eur J Cancer 44 (16): 2461-9, 2008.
- Hagedorn N, Acquaviva C, Fronkova E, et al.: Submicroscopic bone marrow involvement in isolated extramedullary relapses in childhood acute lymphoblastic leukemia: a more precise definition of "isolated" and its possible clinical implications, a collaborative study of the Resistant Disease Committee of the International BFM study group. Blood 110 (12): 4022-9, 2007.
- Ribeiro RC, Rivera GK, Hudson M, et al.: An intensive re-treatment protocol for children with an isolated CNS relapse of acute lymphoblastic leukemia. J Clin Oncol 13 (2): 333-8, 1995.
- Kumar P, Kun LE, Hustu HO, et al.: Survival outcome following isolated central nervous system relapse treated with additional chemotherapy and craniospinal irradiation in childhood acute lymphoblastic leukemia. Int J Radiat Oncol Biol Phys 31 (3): 477-83, 1995.
- Hastings C, Chen Y, Devidas M, et al.: Late isolated central nervous system relapse in childhood B-cell acute lymphoblastic leukemia treated with intensified systemic therapy and delayed reduced dose cranial radiation: A report from the Children's Oncology Group study AALL02P2. Pediatr Blood Cancer 68 (12): e29256, 2021.
- Yoshihara T, Morimoto A, Kuroda H, et al.: Allogeneic stem cell transplantation in children with acute lymphoblastic leukemia after isolated central nervous system relapse: our experiences and review of the literature. Bone Marrow Transplant 37 (1): 25-31, 2006.
- Harker-Murray PD, Thomas AJ, Wagner JE, et al.: Allogeneic hematopoietic cell transplantation in children with relapsed acute lymphoblastic leukemia isolated to the central nervous system. Biol Blood Marrow Transplant 14 (6): 685-92, 2008.
- Eapen M, Zhang MJ, Devidas M, et al.: Outcomes after HLA-matched sibling transplantation or chemotherapy in children with acute lymphoblastic leukemia in a second remission after an isolated central nervous system relapse: a collaborative study of the Children's Oncology Group and the Center for International Blood and Marrow Transplant Research. Leukemia 22 (2): 281-6, 2008.
- Wofford MM, Smith SD, Shuster JJ, et al.: Treatment of occult or late overt testicular relapse in children with acute lymphoblastic leukemia: a Pediatric Oncology Group study. J Clin Oncol 10 (4): 624-30, 1992.
- Trigg ME, Steinherz PG, Chappell R, et al.: Early testicular biopsy in males with acute lymphoblastic leukemia: lack of impact on subsequent event-free survival. J Pediatr Hematol Oncol 22 (1): 27-33, 2000 Jan-Feb.
- van den Berg H, Langeveld NE, Veenhof CH, et al.: Treatment of isolated testicular recurrence of acute lymphoblastic leukemia without radiotherapy. Report from the Dutch Late Effects Study Group. Cancer 79 (11): 2257-62, 1997.
- Barredo JC, Hastings C, Lu X, et al.: Isolated late testicular relapse of B-cell acute lymphoblastic leukemia treated with intensive systemic chemotherapy and response-based testicular radiation: A Children's Oncology Group study. Pediatr Blood Cancer 65 (5): e26928, 2018.
Actualizaciones más recientes a este resumen (07/02/2024)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Características citogenéticas y genómicas de la leucemia linfoblástica aguda infantil
Se añadió a Öfverholm et al. como referencia 149.
Tratamiento de la leucemia linfoblástica aguda infantil recién diagnosticada
Se añadió texto para indicar que en un intento por disminuir las reacciones de hipersensibilidad a la pegaspargasa, en el protocolo Dutch Childhood Oncology Group-ALL11 se asignó al azar a los pacientes para recibir dosis continuas o discontinuas después de la terapia de inducción. En el grupo de dosis continuas, el número de reacciones de hipersensibilidad o inactivación silenciosa fue 7 veces menor y las concentraciones de anticuerpos fueron significativamente inferiores. No hubo diferencias entre los grupos de tratamiento en el número total de efectos tóxicos producidos por la asparaginasa ni en las incidencias a 5 años de recaída, mortalidad y supervivencia sin enfermedad (se citó a Van der Sluis et al. como referencia 26).
Tratamiento de la leucemia linfoblástica aguda infantil en recaída
Se añadió texto para indicar que, dado que los informes preliminares sobre la eficacia de la terapia con células T con receptor de antígeno quimérico (CAR) dirigido al CD19 eran mucho mejores que las experiencias anteriores, los ensayos que derivaron en la aprobación regulatoria del tisagenlecleucel para la LLA con múltiples recaídas o resistencias a tratamientos se llevaron adelante sin aleatorización; su diseño estadístico se basó en experiencias anteriores. En los análisis retrospectivos emparejados con controles en los que se usaron cohortes de tratamiento estándar anteriores a la terapia de células T con CAR se observaron importantes mejoras en la supervivencia general y en la supervivencia sin complicaciones con el uso de tisagenlecleucel. Sin embargo, hasta ahora, en ningún ensayo prospectivo se han comparado los abordajes de células T con CAR y otras modalidades de tratamiento (se citó a V Stackelberg et al. como referencia 177).
El Consejo editorial del PDQ sobre el tratamiento pediátrico es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este resumen del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento de la leucemia linfoblástica aguda infantil. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El consejo editorial del PDQ sobre el tratamiento pediátrico, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
- Si el artículo se debe analizar en una reunión del consejo.
- Si conviene añadir texto acerca del artículo.
- Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Tratamiento de la leucemia linfoblástica aguda infantil son:
- William L. Carroll, MD (Laura and Isaac Perlmutter Cancer Center at NYU Langone)
- Alan Scott Gamis, MD, MPH (Children's Mercy Hospital)
- Michelle Hermiston, MD, PhD (University of California, San Francisco)
- Karen J. Marcus, MD, FACR (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Jessica Pollard, MD (Dana-Farber/Boston Children's Cancer and Blood Disorders Center)
- Michael A. Pulsipher, MD (Huntsman Cancer Institute at University of Utah)
- Rachel E. Rau, MD (University of Washington School of Medicine, Seatle Children’s)
- Arthur Kim Ritchey, MD (Children's Hospital of Pittsburgh of UPMC)
- Lewis B. Silverman, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Malcolm A. Smith, MD, PhD (National Cancer Institute)
- Sarah K. Tasian, MD (Children's Hospital of Philadelphia)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El consejo editorial del PDQ sobre el tratamiento pediátrico emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como “En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]”.
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
PDQ® sobre el tratamiento pediátrico. PDQ Tratamiento de la leucemia linfoblástica aguda infantil. Bethesda, MD: National Cancer Institute. Actualización:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como “estándar” o “en evaluación clínica”. Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Comuníquese con el Instituto Nacional del Cáncer
Para obtener más información sobre las opciones para comunicarse con el NCI, incluso la dirección de correo electrónico, el número telefónico o el chat, consultar la página del Servicio de Información de Cáncer del Instituto Nacional del Cáncer.