Instituto Nacional del Cáncer
Fecha de publicación: Apr 3, 2024
Información sobre el linfoma no Hodgkin (LNH) de células B, su clasificación y opciones de tratamiento: quimioterapia, radiación, terapia dirigida, plasmaféresis, vigilancia, trasplante de células madre y cirugía. Resumen para profesionales de la salud.
Tratamiento del linfoma no Hodgkin de células B
Información general sobre el linfoma no Hodgkin de células B
Los linfomas no Hodgkin (LNH o linfomas no hodgkinianos) son un grupo heterogéneo de neoplasias malignas linfoproliferativas con diferentes modelos de comportamiento y respuestas terapéuticas. Este resumen se centra principalmente en el LNH de células o linfocitos B, que representa alrededor del 85 % de los casos de LNH. Para obtener más información sobre los linfomas de células T, consultar Tratamiento del linfoma no Hodgkin periférico de células T y Tratamiento de la micosis fungoide (incluso el síndrome de Sézary).
Al igual que el linfoma de Hodgkin, el LNH a menudo se origina en los tejidos linfoides y se disemina a otros órganos. Sin embargo, el comportamiento del LNH es mucho menos predecible que el del linfoma de Hodgkin y tiene una tendencia mucho más alta por la diseminación a sitios extraganglionares. El pronóstico depende del tipo histológico, el estadio de la enfermedad y el tratamiento.
Incidencia y mortalidad
Número estimado de casos nuevos y defunciones por LNH en los Estados Unidos en 2024:
- Casos nuevos: 80 620.
- Defunciones: 20 140.
Los linfomas de células B representan alrededor del 85 % de los casos de LNH.
Características anatómicas
El LNH por lo general se origina en los tejidos linfoides.
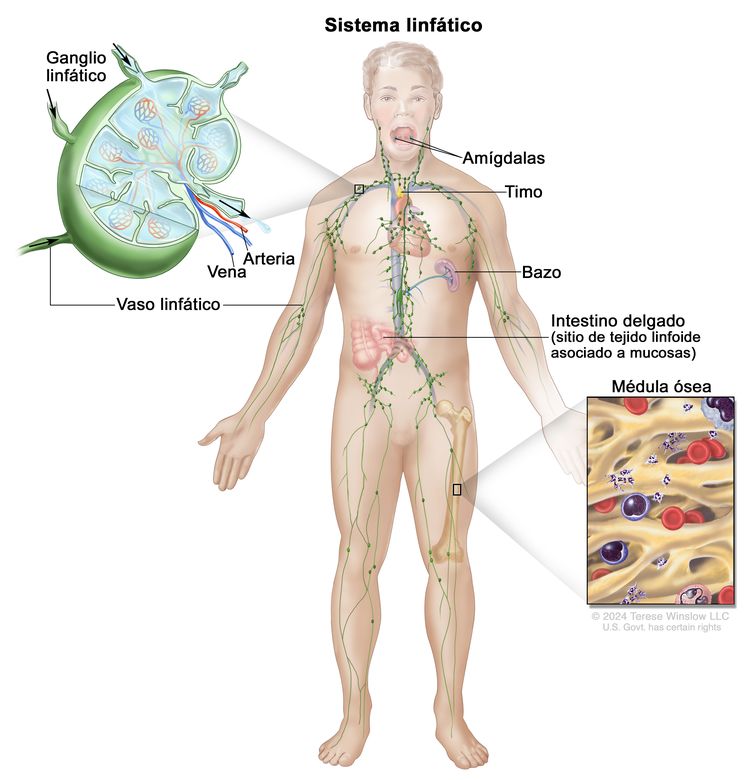 Anatomía del sistema linfático.
Anatomía del sistema linfático.
Pronóstico y supervivencia
El LNH se divide en dos grupos pronósticos: linfomas de crecimiento lento (es decir, de bajo grado de malignidad o indolente) y linfomas de crecimiento rápido (es decir, de alto grado de malignidad o agresivos).
El LNH de crecimiento lento tiene un pronóstico relativamente bueno, con una mediana de supervivencia de hasta 20 años, pero a menudo es incurable en estadios clínicos avanzados. El linfoma no Hodgkin de crecimiento lento en estadios tempranos (estadios I y II) se puede tratar de manera eficaz con radioterapia sola. La mayoría de estos linfomas de crecimiento lento son de tipo morfológico nodular (o folicular).
El LNH de crecimiento rápido tiene una evolución natural más corta, pero un número significativo de estos pacientes se cura con regímenes de quimioterapia combinada intensiva.
Con el tratamiento moderno, la tasa de supervivencia general a 5 años suele ser superior al 60 % en los pacientes con LNH. Más del 50 % de los pacientes con LNH de crecimiento rápido se pueden curar. La mayoría de las recaídas se presentan en los primeros 2 años después del tratamiento. El riesgo de recaída tardía es más alto en los pacientes que manifiestan ambos tipos histológicos: de crecimiento lento (indolente) y de crecimiento rápido (agresivo).
Aunque el LNH de crecimiento lento es sensible a la inmunoterapia, la radioterapia y la quimioterapia, por lo general, se observa una tasa continua de recaída en los estadios avanzados. Sin embargo, a menudo es posible volver a tratar a los pacientes con gran éxito si el tipo histológico de la enfermedad sigue siendo de grado bajo. Los pacientes que presentan al inicio tipos de LNH de crecimiento rápido o cuya enfermedad se convierte en tipos de crecimiento rápido pueden lograr remisiones completas prolongadas con regímenes quimioterapéuticos combinados o consolidación intensiva con apoyo de células madre o médula.
Efectos tardíos del tratamiento del linfoma no Hodgkin
Se han observado efectos tardíos del tratamiento del linfoma no Hodgkin (LNH). Es posible que se presente una alteración de la fertilidad o la capacidad reproductiva después de la exposición a alquilantes. Hasta tres décadas después del diagnóstico, los pacientes tienen un riesgo significativamente elevado de presentar segundos cánceres primarios, en especial los siguientes:
- Cáncer de pulmón.
- Cáncer de encéfalo.
- Cáncer de riñón.
- Cáncer de vejiga.
- Melanoma.
- Linfoma de Hodgkin.
- Leucemia no linfocítica aguda.
La disfunción ventricular izquierda fue un efecto tardío significativo en los sobrevivientes a largo plazo de LNH de grado alto que recibieron más de 200 mg/m² de doxorrubicina.
El síndrome mielodisplásico y la leucemia mielógena aguda son complicaciones tardías de la terapia mielosupresora con apoyo de médula ósea autógena o células madre periféricas, así como de la quimioterapia convencional con alquilantes. La mayoría de estos pacientes presentan hematopoyesis clonal incluso antes del trasplante, lo que indica que la lesión hematológica por lo general se presenta durante la quimioterapia de inducción o reinducción. Se realizó un seguimiento, durante una mediana de 10 años, de una serie de 605 pacientes que recibieron trasplante de médula ósea (TMO) autógeno con ciclofosfamida y radioterapia corporal total (como acondicionamiento). La incidencia de una segunda neoplasia maligna fue del 21 %, y el 10 % de esas neoplasias malignas fueron tumores sólidos.
En un estudio de mujeres jóvenes que recibieron TMO autógeno, se notificaron embarazos exitosos con bebés que nacieron sin anomalías congénitas. La tromboembolia venosa tardía se puede presentar después de un TMO alogénico o autógeno.
Algunos pacientes presentan osteopenia u osteoporosis al inicio del tratamiento; es posible que la densidad ósea empeore después del tratamiento para el linfoma.
En un estudio retrospectivo de cohortes de 21 690 sobrevivientes de linfoma difuso de células B grandes del California Cancer Registry, se evaluó el deterioro a largo plazo de la salud inmunitaria. Se encontraron cocientes de tasas de incidencia elevadas hasta 10 años después para neumonía (10,8 veces más altas), meningitis (5,3 veces más altas), deficiencia de inmunoglobulina (17,6 veces más altas) y citopenias autoinmunitarias (12 veces más altas). De manera similar, hay respuestas humorales alteradas a la vacunación contra el virus SARS-CoV-2 que causa la COVID-19 en pacientes con linfoma que reciben terapias dirigidas a las células B.
References
- Shankland KR, Armitage JO, Hancock BW: Non-Hodgkin lymphoma. Lancet 380 (9844): 848-57, 2012.
- American Cancer Society: Types of B-cell Lymphoma. American Cancer Society, 2019. Available online. Last accessed February 2, 2024.
- American Cancer Society: Cancer Facts and Figures 2024. American Cancer Society, 2024. Available online. Last accessed January 17, 2024.
- Tan D, Horning SJ, Hoppe RT, et al.: Improvements in observed and relative survival in follicular grade 1-2 lymphoma during 4 decades: the Stanford University experience. Blood 122 (6): 981-7, 2013.
- Cabanillas F, Velasquez WS, Hagemeister FB, et al.: Clinical, biologic, and histologic features of late relapses in diffuse large cell lymphoma. Blood 79 (4): 1024-8, 1992.
- Bastion Y, Sebban C, Berger F, et al.: Incidence, predictive factors, and outcome of lymphoma transformation in follicular lymphoma patients. J Clin Oncol 15 (4): 1587-94, 1997.
- Yuen AR, Kamel OW, Halpern J, et al.: Long-term survival after histologic transformation of low-grade follicular lymphoma. J Clin Oncol 13 (7): 1726-33, 1995.
- Haddy TB, Adde MA, McCalla J, et al.: Late effects in long-term survivors of high-grade non-Hodgkin's lymphomas. J Clin Oncol 16 (6): 2070-9, 1998.
- Travis LB, Curtis RE, Glimelius B, et al.: Second cancers among long-term survivors of non-Hodgkin's lymphoma. J Natl Cancer Inst 85 (23): 1932-7, 1993.
- Mudie NY, Swerdlow AJ, Higgins CD, et al.: Risk of second malignancy after non-Hodgkin's lymphoma: a British Cohort Study. J Clin Oncol 24 (10): 1568-74, 2006.
- Hemminki K, Lenner P, Sundquist J, et al.: Risk of subsequent solid tumors after non-Hodgkin's lymphoma: effect of diagnostic age and time since diagnosis. J Clin Oncol 26 (11): 1850-7, 2008.
- Major A, Smith DE, Ghosh D, et al.: Risk and subtypes of secondary primary malignancies in diffuse large B-cell lymphoma survivors change over time based on stage at diagnosis. Cancer 126 (1): 189-201, 2020.
- Moser EC, Noordijk EM, van Leeuwen FE, et al.: Long-term risk of cardiovascular disease after treatment for aggressive non-Hodgkin lymphoma. Blood 107 (7): 2912-9, 2006.
- Darrington DL, Vose JM, Anderson JR, et al.: Incidence and characterization of secondary myelodysplastic syndrome and acute myelogenous leukemia following high-dose chemoradiotherapy and autologous stem-cell transplantation for lymphoid malignancies. J Clin Oncol 12 (12): 2527-34, 1994.
- Stone RM, Neuberg D, Soiffer R, et al.: Myelodysplastic syndrome as a late complication following autologous bone marrow transplantation for non-Hodgkin's lymphoma. J Clin Oncol 12 (12): 2535-42, 1994.
- Armitage JO, Carbone PP, Connors JM, et al.: Treatment-related myelodysplasia and acute leukemia in non-Hodgkin's lymphoma patients. J Clin Oncol 21 (5): 897-906, 2003.
- André M, Mounier N, Leleu X, et al.: Second cancers and late toxicities after treatment of aggressive non-Hodgkin lymphoma with the ACVBP regimen: a GELA cohort study on 2837 patients. Blood 103 (4): 1222-8, 2004.
- Oddou S, Vey N, Viens P, et al.: Second neoplasms following high-dose chemotherapy and autologous stem cell transplantation for malignant lymphomas: a report of six cases in a cohort of 171 patients from a single institution. Leuk Lymphoma 31 (1-2): 187-94, 1998.
- Lenz G, Dreyling M, Schiegnitz E, et al.: Moderate increase of secondary hematologic malignancies after myeloablative radiochemotherapy and autologous stem-cell transplantation in patients with indolent lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group. J Clin Oncol 22 (24): 4926-33, 2004.
- McLaughlin P, Estey E, Glassman A, et al.: Myelodysplasia and acute myeloid leukemia following therapy for indolent lymphoma with fludarabine, mitoxantrone, and dexamethasone (FND) plus rituximab and interferon alpha. Blood 105 (12): 4573-5, 2005.
- Morton LM, Curtis RE, Linet MS, et al.: Second malignancy risks after non-Hodgkin's lymphoma and chronic lymphocytic leukemia: differences by lymphoma subtype. J Clin Oncol 28 (33): 4935-44, 2010.
- Mach-Pascual S, Legare RD, Lu D, et al.: Predictive value of clonality assays in patients with non-Hodgkin's lymphoma undergoing autologous bone marrow transplant: a single institution study. Blood 91 (12): 4496-503, 1998.
- Lillington DM, Micallef IN, Carpenter E, et al.: Detection of chromosome abnormalities pre-high-dose treatment in patients developing therapy-related myelodysplasia and secondary acute myelogenous leukemia after treatment for non-Hodgkin's lymphoma. J Clin Oncol 19 (9): 2472-81, 2001.
- Brown JR, Yeckes H, Friedberg JW, et al.: Increasing incidence of late second malignancies after conditioning with cyclophosphamide and total-body irradiation and autologous bone marrow transplantation for non-Hodgkin's lymphoma. J Clin Oncol 23 (10): 2208-14, 2005.
- Jackson GH, Wood A, Taylor PR, et al.: Early high dose chemotherapy intensification with autologous bone marrow transplantation in lymphoma associated with retention of fertility and normal pregnancies in females. Scotland and Newcastle Lymphoma Group, UK. Leuk Lymphoma 28 (1-2): 127-32, 1997.
- Gangaraju R, Chen Y, Hageman L, et al.: Risk of venous thromboembolism in patients with non-Hodgkin lymphoma surviving blood or marrow transplantation. Cancer 125 (24): 4498-4508, 2019.
- Westin JR, Thompson MA, Cataldo VD, et al.: Zoledronic acid for prevention of bone loss in patients receiving primary therapy for lymphomas: a prospective, randomized controlled phase III trial. Clin Lymphoma Myeloma Leuk 13 (2): 99-105, 2013.
- Shree T, Li Q, Glaser SL, et al.: Impaired Immune Health in Survivors of Diffuse Large B-Cell Lymphoma. J Clin Oncol 38 (15): 1664-1675, 2020.
- Ghione P, Gu JJ, Attwood K, et al.: Impaired humoral responses to COVID-19 vaccination in patients with lymphoma receiving B-cell-directed therapies. Blood 138 (9): 811-814, 2021.
- Terpos E, Trougakos IP, Gavriatopoulou M, et al.: Low neutralizing antibody responses against SARS-CoV-2 in older patients with myeloma after the first BNT162b2 vaccine dose. Blood 137 (26): 3674-3676, 2021.
Clasificación celular del linfoma no Hodgkin de células B
Se debe consultar con un patólogo antes de realizar una biopsia porque algunos estudios exigen una preparación especial del tejido (por ejemplo, tejido congelado). El conocimiento de los marcadores de superficie celular y los reordenamientos de los genes de inmunoglobulinas y del receptor de células T puede ser útil al momento de tomar decisiones diagnósticas y terapéuticas. El exceso clonal de cadenas ligeras de inmunoglobulina permite diferenciar las células linfoides malignas de las células reactivas. Es muy importante que un hematopatólogo con experiencia en el diagnóstico de linfomas realice un examen minucioso de las muestras de biopsia externas porque las características histopatológicas afectan el pronóstico y el abordaje del tratamiento. Aunque se recomienda obtener biopsias de ganglios linfáticos cuando sea posible, a veces los datos inmunofenotípicos son suficientes para establecer un diagnóstico de linfoma cuando es preferible hacer un estudio citológico mediante aspiración con aguja fina o biopsia con aguja gruesa.
Sistemas de clasificación tradicionales
Históricamente, no se había podido establecer un tratamiento uniforme para los pacientes con linfoma no Hodgkin (LNH) por la falta de un sistema de clasificación unificado. En 1982, se publicaron los resultados de un estudio de consenso llamado Working Formulation. En el Working Formulation se combinaron los resultados de 6 sistemas de clasificación principales para formar una clasificación. Esto permitió la comparación de estudios de diferentes instituciones y países. La clasificación de Rappaport, que también aparece a continuación, ya no se usa mucho.
| Working Formulation | Clasificación de Rappaport |
|---|---|
| Grado bajo | |
| A. Linfocítico pequeño, compatible con leucemia linfocítica crónica | Linfocítico difuso, bien diferenciado |
| B. Folicular con predominio de células hendidas pequeñas | Linfocítico nodular, poco diferenciado (pobremente diferenciado) |
| C. Folicular mixto con mezcla de células hendidas pequeñas y células grandes | Nodular mixto, linfocítico e histiocítico |
| Grado intermedio | |
| D. Folicular con predominio de células grandes | Histiocítico nodular |
| E. Difuso con células hendidas pequeñas | Linfocítico difuso, poco diferenciado (pobremente diferenciado) |
| F. Difuso mixto con células pequeñas y grandes | Difuso mixto, linfocítico e histiocítico |
| G. Difuso con células grandes hendidas o no hendidas | Histiocítico difuso |
| Grado alto | |
| H. Inmunoblástico con células grandes | Histiocítico difuso |
| I. Linfoblástico con células cerebriformes o no cerebriformes | Linfoblástico difuso |
| J. Linfoma de Burkitt o no Burkitt con células pequeñas no hendidas | Indiferenciado difuso de Burkitt o no Burkitt |
Sistemas de clasificación actuales
A medida que el diagnóstico histopatológico del LNH es más sofisticado gracias a las técnicas inmunológicas y genéticas, se describen varias entidades patológicas nuevas. Además, la comprensión y el tratamiento de muchos de los subtipos patológicos descritos antes han cambiado. Como resultado, el sistema de clasificación Working Formulation es obsoleto y carece de utilidad para profesionales clínicos y patólogos. Por este motivo los patólogos europeos y norteamericanos propusieron una clasificación nueva: la clasificación europea americana revisada del linfoma (Revised European American Lymphoma), conocida como clasificación REAL. Desde 1995, los miembros de la European Association for Haematopathology y de la Society for Hematopathology colaboran para la formulación de una clasificación nueva de la Organización Mundial de la Salud (OMS), que representa una versión actualizada del sistema REAL.
Clasificación REAL modificada por la Organización Mundial de la Salud
En la modificación que la Organización Mundial de la Salud (OMS) hizo a la clasificación europea americana revisada del linfoma (Revised European American Lymphoma [REAL]) se reconocen tres categorías principales de neoplasias linfoides malignas según la morfología y el linaje celular: neoplasias de células B, neoplasias de células T o de células citolíticas naturales (NK), y linfoma de Hodgkin (LH). En esta clasificación, se incluyen los linfomas y las leucemias linfoides porque las fases sólidas y circulantes de estas enfermedades se encuentran en muchas neoplasias linfoides, así que la distinción entre ellas es artificial. Por ejemplo, la leucemia linfocítica crónica (LLC) de células B y el linfoma linfocítico de células B pequeñas son distintas manifestaciones de la misma neoplasia, al igual que los linfomas linfoblásticos y las leucemias linfocíticas agudas. Dentro de las categorías de células B y células T, se reconocen dos subdivisiones: neoplasias precursoras, que corresponden a los estadios más tempranos de diferenciación, y neoplasias diferenciadas más maduras.
- Neoplasias de células B precursoras: leucemia linfoblástica aguda de células B precursoras o linfoma linfoblástico de células B precursoras.
- Neoplasias de células B periféricas.
- Leucemia linfocítica crónica de células B o linfoma linfocítico de células B pequeñas.
- Leucemia prolinfocítica de células B.
- Linfoma linfoplasmocítico o inmunocitoma.
- Linfoma de células de manto.
- Linfoma folicular.
- Linfoma extraganglionar de células B de zona marginal de tejido linfoide asociado a mucosas.
- Linfoma ganglionar de células B de zona marginal (± células B monocitoides).
- Linfoma esplénico de zona marginal (± linfocitos vellosos).
- Leucemia de células pilosas.
- Plasmocitoma o mieloma de células plasmáticas.
- Linfoma difuso de células B grandes.
- Linfoma de Burkitt.
- Neoplasias de células T precursoras: leucemia linfoblástica aguda de células T precursoras o linfoma linfoblástico de células T precursoras. Para obtener más información, consultar Tratamiento de la leucemia linfoblástica aguda en adultos.
- Neoplasias periféricas de células T-NK.
- Leucemia linfocítica crónica de células T o leucemia prolinfocítica de células T.
- Leucemia linfocítica granular de células T.
- Micosis fungoide (incluso el síndrome de Sézary).
- Linfoma periférico de células T sin otra indicación.
- Linfoma hepatoesplénico de células T γ-δ.
- Linfoma subcutáneo de células T similar a la paniculitis.
- Linfoma angioinmunoblástico de células T.
- Linfoma extraganglionar de células T-NK de tipo nasal.
- Linfoma de células T asociado a enteropatía.
- Leucemia o linfoma de células T en adultos (virus linfotrópico humano de células T de tipo 1 [VLHT] 1+).
- Linfoma anaplásico de células grandes de tipo sistémico primario.
- Linfoma anaplásico de células grandes de tipo cutáneo primario.
- Leucemia de células NK de crecimiento rápido (agresiva).
- Linfoma de Hodgkin con predominio linfocítico nodular.
- Linfoma de Hodgkin clásico.
- Linfoma de Hodgkin con esclerosis nodular.
- Linfoma de Hodgkin clásico rico en linfocitos.
- Linfoma de Hodgkin con celularidad mixta.
- Linfoma de Hodgkin con agotamiento linfocítico.
La clasificación REAL abarca todas las neoplasias linfoproliferativas. Para obtener más información, consultar los siguientes resúmenes del PDQ:
- Tratamiento de la leucemia linfoblástica aguda en adultos
- Tratamiento del linfoma de Hodgkin
- Tratamiento del linfoma relacionado con el SIDA
- Tratamiento de la leucemia linfocítica crónica
- Tratamiento de la leucemia de células pilosas
- Tratamiento de la micosis fungoide (incluso el síndrome de Sézary)
- Tratamiento de las neoplasias de células plasmáticas (incluso mieloma múltiple)
- Tratamiento del linfoma primario del sistema nervioso central
References
- Zeppa P, Marino G, Troncone G, et al.: Fine-needle cytology and flow cytometry immunophenotyping and subclassification of non-Hodgkin lymphoma: a critical review of 307 cases with technical suggestions. Cancer 102 (1): 55-65, 2004.
- Young NA, Al-Saleem T: Diagnosis of lymphoma by fine-needle aspiration cytology using the revised European-American classification of lymphoid neoplasms. Cancer 87 (6): 325-45, 1999.
- National Cancer Institute sponsored study of classifications of non-Hodgkin's lymphomas: summary and description of a working formulation for clinical usage. The Non-Hodgkin's Lymphoma Pathologic Classification Project. Cancer 49 (10): 2112-35, 1982.
- Pugh WC: Is the working formulation adequate for the classification of the low grade lymphomas? Leuk Lymphoma 10 (Suppl 1): 1-8, 1993.
- Harris NL, Jaffe ES, Stein H, et al.: A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 84 (5): 1361-92, 1994.
- Pittaluga S, Bijnens L, Teodorovic I, et al.: Clinical analysis of 670 cases in two trials of the European Organization for the Research and Treatment of Cancer Lymphoma Cooperative Group subtyped according to the Revised European-American Classification of Lymphoid Neoplasms: a comparison with the Working Formulation. Blood 87 (10): 4358-67, 1996.
- Armitage JO, Weisenburger DD: New approach to classifying non-Hodgkin's lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin's Lymphoma Classification Project. J Clin Oncol 16 (8): 2780-95, 1998.
- A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin's lymphoma. The Non-Hodgkin's Lymphoma Classification Project. Blood 89 (11): 3909-18, 1997.
- Pileri SA, Milani M, Fraternali-Orcioni G, et al.: From the R.E.A.L. Classification to the upcoming WHO scheme: a step toward universal categorization of lymphoma entities? Ann Oncol 9 (6): 607-12, 1998.
- Society for Hematopathology Program: Society for Hematopathology Program. Am J Surg Pathol 21 (1): 114-121, 1997.
Información sobre los estadios del linfoma no Hodgkin de células B
El estadio es importante para la selección del tratamiento en pacientes con linfoma no Hodgkin (LNH). Por lo general, las tomografías computarizadas (TC) del tórax y el abdomen forman parte de la evaluación para la estadificación de todos los pacientes con linfoma. El sistema de estadificación para el LNH es similar al que se usa para el linfoma de Hodgkin (LH).
Es común que los pacientes con LNH presenten compromiso de los siguientes sitios:
- Ganglios linfáticos no adyacentes.
- Anillo de Waldeyer.
- Ganglios epitrocleares.
- Tubo digestivo.
- Sitios extraganglionares. (En ocasiones, una localización extraganglionar es el único sitio comprometido en pacientes con un linfoma difuso).
- Médula ósea.
- Hígado (muy común en pacientes con linfomas de grado bajo).
El examen citológico del líquido cefalorraquídeo a veces da un resultado positivo en pacientes con LNH de crecimiento rápido. El compromiso de los ganglios linfáticos hiliares y mediastínicos es menos común que en los pacientes con LH. Sin embargo, las adenopatías mediastínicas son una característica destacada del linfoma linfoblástico y del linfoma mediastínico primario de células B, que se presentan sobre todo en adultos jóvenes.
La mayoría de los pacientes con LNH consultan con una enfermedad avanzada (estadio III o IV) que a menudo se identifica mediante tomografías computarizadas (TC) o biopsias de la médula ósea y de otros sitios comprometidos accesibles. En una revisión retrospectiva de más de 32 000 casos de linfoma en Francia, hasta el 40 % de los diagnósticos se confirmaron mediante biopsia con aguja gruesa y el 60 % mediante biopsia por escisión. Después de la revisión por expertos, la biopsia con aguja gruesa proporcionó un diagnóstico definitivo en el 92,3 % de los casos; mientras que la biopsia por escisión proporcionó un diagnóstico definitivo en el 98,1 % de los casos (P< 0,0001). Por lo general, no se requiere una biopsia laparoscópica o laparotomía para la estadificación, pero es posible que en raras ocasiones se necesite para establecer un diagnóstico o determinar el tipo histológico del linfoma.
La tomografía por emisión de positrones (TEP) con flúor F 18-fludesoxiglucosa se puede usar para la estadificación inicial. También quizás se utilice durante el seguimiento después del tratamiento como complemento de la TC. En múltiples estudios se demostró que las TEP intermedias después de 2 a 4 ciclos de terapia no aportan información pronóstica confiable. En un ensayo de grupo cooperativo grande (ECOG-E344 [NCT00274924]) se notificaron problemas de reproducibilidad entre los observadores. En 2 ensayos prospectivos y en 1 metanálisis no se observaron diferencias en los desenlaces entre los pacientes con resultados negativos en la TEP y los pacientes con resultados positivos en la TEP pero que tenían resultados negativos en la biopsia.
En un estudio retrospectivo de 130 pacientes con linfoma difuso de células B grandes, la TEP permitió identificar el compromiso de la médula ósea con importancia clínica; la biopsia de médula ósea no llevó a la sobreestadificación de ningún paciente con linfoma. En un estudio retrospectivo de 580 pacientes con linfoma folicular de 7 ensayos patrocinados por el Instituto Nacional del Cáncer, no se observó ninguna mejora en la evaluación de la respuesta al tratamiento cuando se añadió la biopsia de la médula ósea a las imágenes radiológicas. La evaluación del LNH debe incluir una biopsia de la médula ósea cuando el resultado posiblemente conlleve un cambio en el tratamiento (por ejemplo, si se encuentra un estadio limitado vs. uno avanzado) o durante la evaluación de la presencia de citopenias.
En los pacientes con linfoma folicular, un resultado positivo para compromiso en una TEP después del tratamiento conlleva un pronóstico más precario; sin embargo, no está claro si un resultado positivo en una TEP es predictivo cuando se implementa un tratamiento adicional o diferente.
Sistema de estadificación
Clasificación de Lugano
El American Joint Committee on Cancer (AJCC) adoptó la clasificación de Lugano para evaluar y estadificar el linfoma. El sistema de clasificación de Lugano reemplazó el sistema de clasificación de Ann Arbor, que se adoptó en 1971 en la conferencia de Ann Arbor, y que fue modificado 18 años después en la reunión de Cotswolds.
| Estadio | Descripción del estadio | Imagen |
|---|---|---|
| LCR = líquido cefalorraquídeo; TC = tomografía computarizada; LDCBG = linfoma difuso de células B grandes; LNH = linfoma no Hodgkin. | ||
| aHodgkin and Non-Hodgkin Lymphomas. En: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8.ª edición Nueva York, NY: Springer, 2017, pp. 937-58. | ||
| bEl estadio II con gran masa tumoral (tumor voluminoso) quizás se considere un estadio temprano o avanzado en función de las características histológicas y los factores pronósticos del linfoma. | ||
| cLa definición de una gran masa tumoral varía según las características histológicas del linfoma. En la clasificación de Lugano, una gran masa tumoral en el linfoma de Hodgkin se define como una masa que mide más de un tercio del diámetro torácico en la TC del tórax, o una masa que mide >10 cm. Las definiciones recomendadas para una gran masa tumoral en el LNH varían de acuerdo a las características histológicas del linfoma. En el linfoma folicular, se sugirió la medida de 6 cm a partir del Follicular Lymphoma International Prognostic Index-2 y su validación. En el LDCBG, se han usado valores de corte que oscilan entre 5 cm y 10 cm, aunque el límite recomendado es de 10 cm. | ||
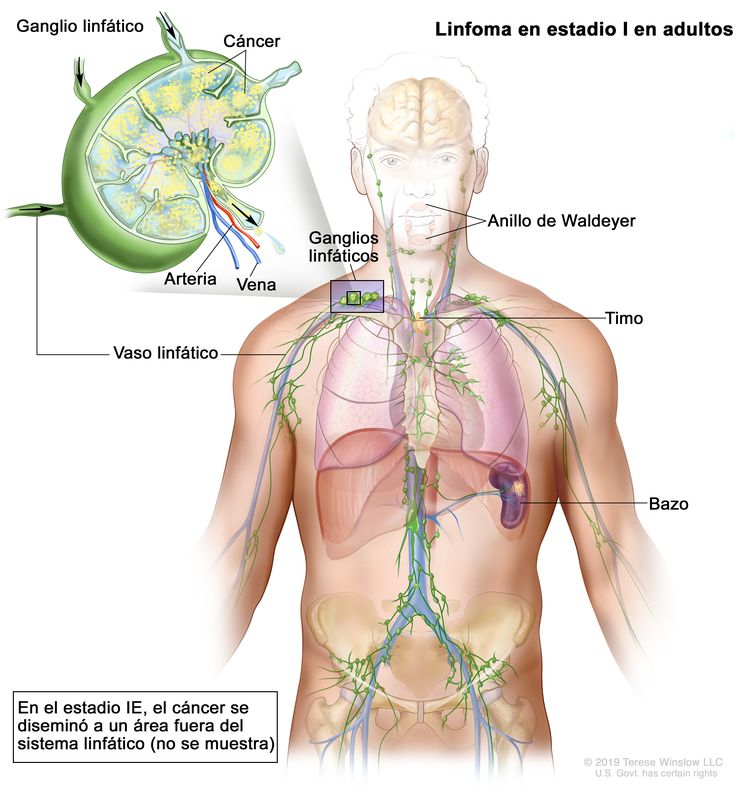
| Estadio limitado | ||
| I | Compromiso de un solo sitio linfático (es decir, una región ganglionar, el anillo de Waldeyer, el timo o el bazo). |
|
| IE | Compromiso de 1 solo sitio extralinfático sin compromiso ganglionar (infrecuente en el linfoma de Hodgkin). | |
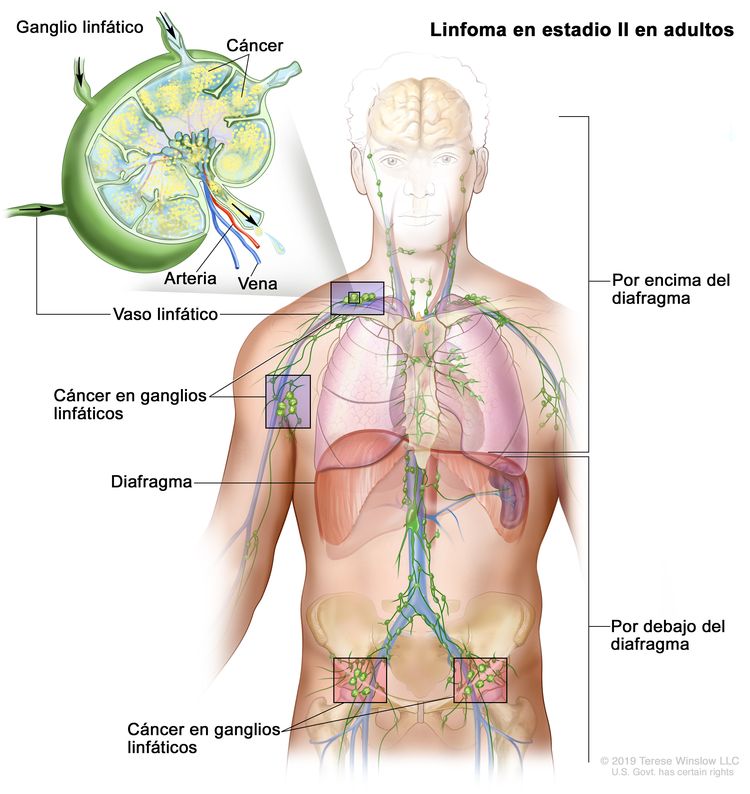
| II | Compromiso de 2 o más regiones ganglionares en el mismo lado del diafragma. |
|
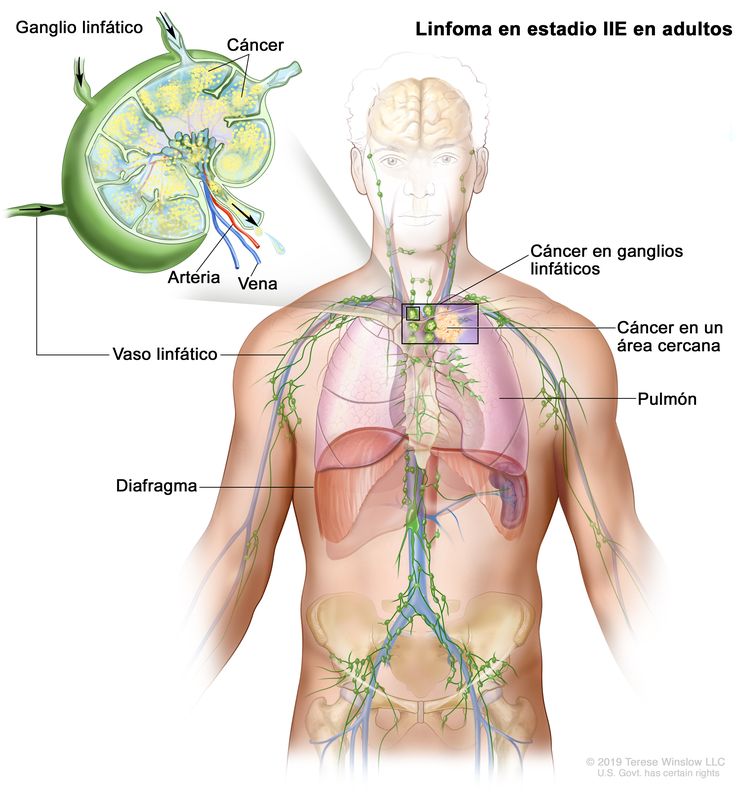
| IIE | Diseminación extralinfática adyacente desde un sitio ganglionar con compromiso de otras regiones ganglionares en el mismo lado del diafragma o sin este. |
|
| II con gran masa tumoralb | Estadio II con gran masa tumoralc | |
| Estadio avanzado | ||
| III | Compromiso de regiones ganglionares en ambos lados del diafragma; adenopatías por encima del diafragma y compromiso esplénico. |
|
| IV | Compromiso difuso o diseminación a uno o más órganos extralinfáticos con compromiso ganglionar o sin este; compromiso de órgano extralinfático no adyacente con enfermedad ganglionar en estadio II; o compromiso de cualquier órgano extralinfático con enfermedad ganglionar en estadio III. El estadio IV incluye cualquier tipo de compromiso del LCR, la médula ósea y el hígado, o la presencia de lesiones pulmonares múltiples (diferentes a las lesiones por diseminación directa de una enfermedad en estadio IIE). |
|
| Nota: Se utiliza la designación A o B junto con el grupo de estadio para el linfoma de Hodgkin. En el LNH ya no se utiliza la designación A ni B. | ||
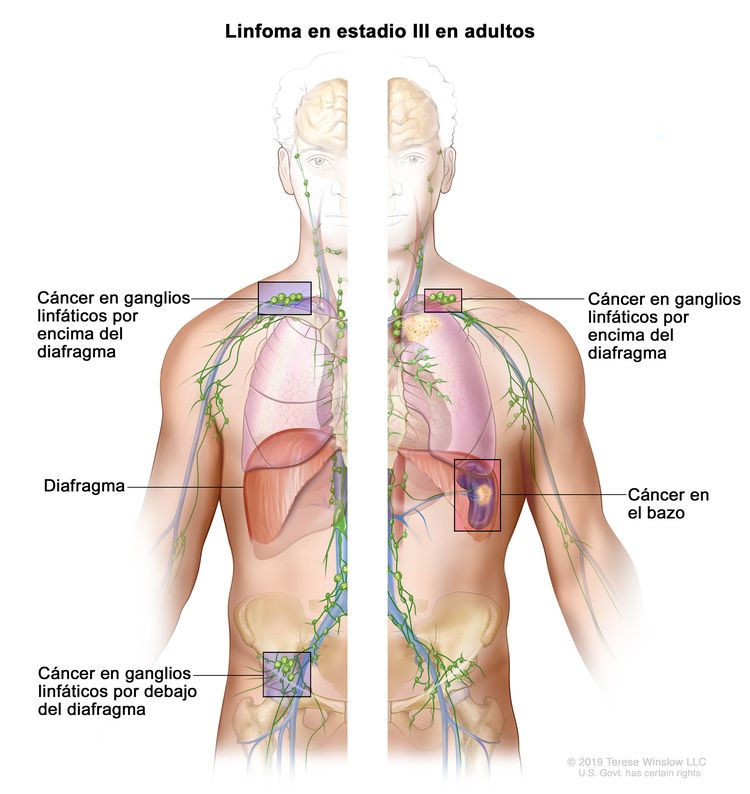
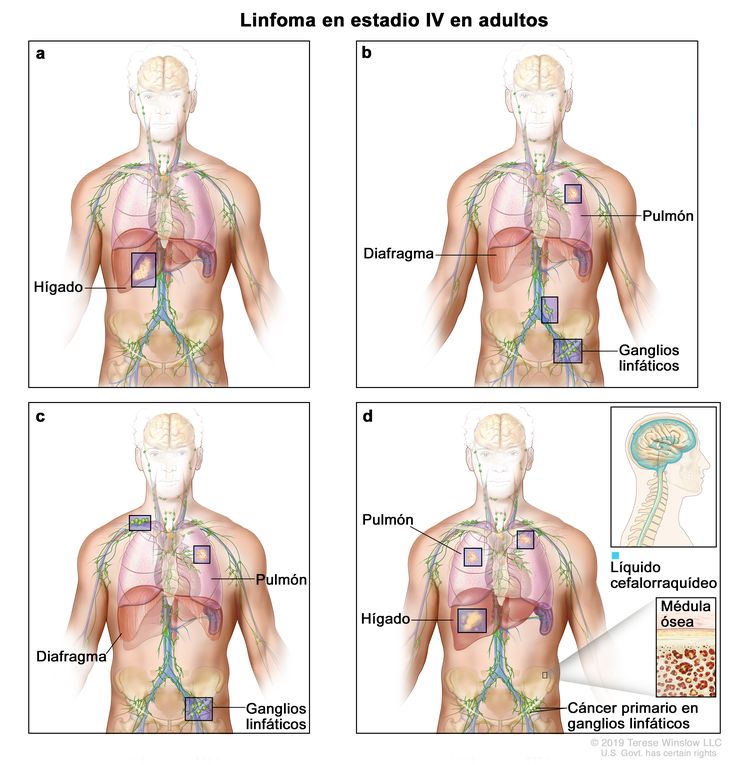
En ocasiones, se usan otros sistemas de estadificación especializados. El médico debe estar al tanto del sistema que se usa en el informe del paciente.
La designación E se usa cuando aparecen neoplasias linfoides extraganglionares malignas en tejidos separados de los conglomerados linfáticos principales pero cercanos a estos. El estadio IV indica una enfermedad con diseminación difusa por todo un sitio extraganglionar, como el hígado. Si el compromiso de uno o más sitios extralinfáticos se documentó mediante estudio patológico, se usa el símbolo del sitio comprometido seguido por el signo más (+).
| N = ganglios | H = hígado | L = pulmón | M = médula ósea |
| S = bazo | P = pleura | O = hueso | D = piel |
En la práctica actual se asigna un estadio clínico a partir de los hallazgos de la evaluación clínica y un estadio patológico a partir de los hallazgos de los procedimientos invasivos adicionales a la biopsia inicial.
Por ejemplo, es posible que se encuentre un compromiso del hígado y la médula ósea mediante una biopsia percutánea en un paciente con adenopatía inguinal sin síntomas sistémicos que tiene un resultado positivo en el linfangiograma. El estadio exacto para dicho paciente sería estadio clínico IIA, estadio patológico IVA(H+)(M+).
Hay otros factores que no se incluyen en el sistema de estadificación anterior, pero que son importantes para la estadificación y el pronóstico de los pacientes con LNH. Estos factores son los siguientes:
- Edad.
- Estado funcional (EF).
- Tamaño del tumor.
- Concentraciones de lactato–deshidrogenasa (LDH).
- Número de sitios con compromiso extraganglionar.
En el índice pronóstico internacional de la National Comprehensive Cancer Network (National Comprehensive Cancer Network International Prognostic Index [NCCN-IPI]) para el linfoma no Hodgkin (LNH) de crecimiento rápido (linfoma difuso de células grandes) se identifican los siguientes 5 factores de riesgo significativos para el pronóstico de la supervivencia general (SG) y los puntajes de riesgo relacionado:
- Edad.
- <40 años: 0.
- 41–60 años: 1.
- 61–75 años: 2.
- >75 años: 3.
- Estadio III o IV: 1.
- Estado funcional (EF) 2, 3 o 4: 1.
- Concentración sérica de lactato–deshidrogenasa (LDH).
- Normalizada: 0.
- >1x–3x: 1.
- >3x: 2.
- Número de sitios extraganglionares ≥2: 1.
Puntajes de riesgo:
- Bajo (0 o 1): Tasa de SG a 5 años, 96 %; tasa de supervivencia sin progresión (SSP), 91 %.
- Intermedio bajo (2 o 3): Tasa de SG a 5 años, 82 %; tasa de SSP, 74 %.
- Intermedio alto (4 o 5): Tasa de SG a 5 años, 64 %; tasa de SSP, 51 %.
- Alto (>6): Tasa de SG a 5 años, 33 %; tasa de SSP, 30 %.
Se utilizan modificaciones del IPI ajustadas por edad y estadio para los pacientes más jóvenes con enfermedad localizada. Los intervalos de tiempo más cortos entre el diagnóstico y el tratamiento son marcadores indirectos de factores biológicos de pronóstico precario.
El gen BCL2 y el reordenamiento del gen MYC o la sobreexpresión dual del gen MYC, o ambos, confieren un pronóstico particularmente precario. Los pacientes con riesgo alto de recaída quizás se beneficien de terapia de consolidación u otros abordajes en evaluación clínica. La obtención de perfiles moleculares de expresión génica mediante micromatrices de ADN quizás sirva en el futuro para ayudar a estratificar a los pacientes en grupos de tratamiento dirigido a dianas específicas y para mejorar la predicción de la supervivencia después de la quimioterapia estándar.
References
- Syrykh C, Chaouat C, Poullot E, et al.: Lymph node excisions provide more precise lymphoma diagnoses than core biopsies: a French Lymphopath network survey. Blood 140 (24): 2573-2583, 2022.
- Mann GB, Conlon KC, LaQuaglia M, et al.: Emerging role of laparoscopy in the diagnosis of lymphoma. J Clin Oncol 16 (5): 1909-15, 1998.
- Barrington SF, Mikhaeel NG, Kostakoglu L, et al.: Role of imaging in the staging and response assessment of lymphoma: consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol 32 (27): 3048-58, 2014.
- Horning SJ, Juweid ME, Schöder H, et al.: Interim positron emission tomography scans in diffuse large B-cell lymphoma: an independent expert nuclear medicine evaluation of the Eastern Cooperative Oncology Group E3404 study. Blood 115 (4): 775-7; quiz 918, 2010.
- Moskowitz CH, Schöder H, Teruya-Feldstein J, et al.: Risk-adapted dose-dense immunochemotherapy determined by interim FDG-PET in Advanced-stage diffuse large B-Cell lymphoma. J Clin Oncol 28 (11): 1896-903, 2010.
- Pregno P, Chiappella A, Bellò M, et al.: Interim 18-FDG-PET/CT failed to predict the outcome in diffuse large B-cell lymphoma patients treated at the diagnosis with rituximab-CHOP. Blood 119 (9): 2066-73, 2012.
- Sun N, Zhao J, Qiao W, et al.: Predictive value of interim PET/CT in DLBCL treated with R-CHOP: meta-analysis. Biomed Res Int 2015: 648572, 2015.
- Khan AB, Barrington SF, Mikhaeel NG, et al.: PET-CT staging of DLBCL accurately identifies and provides new insight into the clinical significance of bone marrow involvement. Blood 122 (1): 61-7, 2013.
- Rutherford SC, Yin J, Pederson L, et al.: Relevance of Bone Marrow Biopsies for Response Assessment in US National Cancer Institute National Clinical Trials Network Follicular Lymphoma Clinical Trials. J Clin Oncol 41 (2): 336-342, 2023.
- Pyo J, Won Kim K, Jacene HA, et al.: End-therapy positron emission tomography for treatment response assessment in follicular lymphoma: a systematic review and meta-analysis. Clin Cancer Res 19 (23): 6566-77, 2013.
- Hodgkin and non-Hodgkin lymphoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp. 937–58.
- Carbone PP, Kaplan HS, Musshoff K, et al.: Report of the Committee on Hodgkin's Disease Staging Classification. Cancer Res 31 (11): 1860-1, 1971.
- Lister TA, Crowther D, Sutcliffe SB, et al.: Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin's disease: Cotswolds meeting. J Clin Oncol 7 (11): 1630-6, 1989.
- National Cancer Institute sponsored study of classifications of non-Hodgkin's lymphomas: summary and description of a working formulation for clinical usage. The Non-Hodgkin's Lymphoma Pathologic Classification Project. Cancer 49 (10): 2112-35, 1982.
- Zhou Z, Sehn LH, Rademaker AW, et al.: An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 123 (6): 837-42, 2014.
- Møller MB, Christensen BE, Pedersen NT: Prognosis of localized diffuse large B-cell lymphoma in younger patients. Cancer 98 (3): 516-21, 2003.
- Maurer MJ, Ghesquières H, Link BK, et al.: Diagnosis-to-Treatment Interval Is an Important Clinical Factor in Newly Diagnosed Diffuse Large B-Cell Lymphoma and Has Implication for Bias in Clinical Trials. J Clin Oncol 36 (16): 1603-1610, 2018.
- Scott DW, King RL, Staiger AM, et al.: High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with diffuse large B-cell lymphoma morphology. Blood 131 (18): 2060-2064, 2018.
- Horn H, Ziepert M, Becher C, et al.: MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 121 (12): 2253-63, 2013.
- A predictive model for aggressive non-Hodgkin's lymphoma. The International Non-Hodgkin's Lymphoma Prognostic Factors Project. N Engl J Med 329 (14): 987-94, 1993.
- Sha C, Barrans S, Cucco F, et al.: Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. J Clin Oncol 37 (3): 202-212, 2019.
Linfoma no Hodgkin de células B de crecimiento lento
El linfoma no Hodgkin (LNH) de crecimiento lento (indolente) incluye los siguientes subtipos:
- Linfoma folicular (grados 1–3a).
- Linfoma linfoplasmocítico (macroglobulinemia de Waldenström).
- Linfoma de zona marginal.
Linfoma folicular (grados 1–3a)
El linfoma folicular representa el 20 % de todos los LNH y hasta el 70 % de los linfomas de crecimiento lento notificados en ensayos clínicos norteamericanos y europeos. La mayoría de los pacientes con linfoma folicular tienen 50 años o más y exhiben una enfermedad diseminada en el momento del diagnóstico. El compromiso ganglionar es más común y, con frecuencia, se acompaña de enfermedad en el bazo y la médula ósea. El reordenamiento del gen BCL2 está presente en más del 90 % de los pacientes con linfoma folicular. La sobreexpresión de la proteína BCL2 se relaciona con la incapacidad de erradicar el linfoma mediante la inhibición de la apoptosis.
Pronóstico
El linfoma folicular se designa como de crecimiento lento porque la mediana de supervivencia oscila entre 8 y 15 años, incluso en estadios avanzados. Los pacientes con linfoma folicular en estadio avanzado no se curan con las opciones terapéuticas vigentes. La tasa de recaída es bastante constante en el tiempo, aun en pacientes que lograron respuestas completas al tratamiento. La observación cautelosa y el aplazamiento del tratamiento hasta que el paciente presente síntomas, es una opción para los pacientes con linfoma folicular en estadio avanzado. En un índice internacional de linfoma folicular (Follicular Lymphoma International Prognostic Index [FLIPI]) se identificaron 5 factores de riesgo significativos para el pronóstico de la supervivencia general (SG):
- Edad (≤60 vs. >60 años).
- Concentración sérica (normal vs. elevada) de lactato–deshidrogenasa (LDH).
- Estadio (estadio I o II vs. estadio III o IV).
- Concentración de hemoglobina (≥120 g/l vs. <120 g/l).
- Número de áreas ganglionares (≤4 vs. >4).
La tasa de supervivencia a 10 años para los pacientes con menos de 2 factores de riesgo es del 85 %, y para aquellos con 3 o más factores de riesgo es del 40 %. En los criterios FLIPI-2 revisados, se propuso que la concentración elevada de microglobulina β2 y el tamaño de un ganglio linfático de más de 6 cm fueran los factores pronósticos en lugar de la concentración sérica de LDH y el número de áreas ganglionares. Aunque los índices FLIPI y FLIPI-2 pueden predecir la supervivencia sin progresión (SSP) y la SG, no es posible usar los puntajes para establecer la necesidad de tratamiento ni para predecir la respuesta terapéutica. La principal utilidad de FLIPI y FLIPI-2 es garantizar el equilibrio de los factores pronósticos o definir los requisitos para participar en ensayos clínicos aleatorizados. Las personas con un puntaje de FLIPI adverso quizás se beneficien de una observación cautelosa o quizás respondan bien al tratamiento inicial. Un índice pronóstico alternativo en el que solo se usan la microglobulina ß2 y el compromiso inicial de la médula ósea (PRIMA-PI) tiene la desventaja de que sea necesario hacer una prueba invasiva que, por lo general, no se requiere fuera del contexto de un ensayo clínico. Un índice pronóstico alternativo que solo utilizó variables clínicas no invasivas superó al FLIPI, FLIPI-2 y PRIMA-PI, con el uso de datos de ensayos de inmunoquimioterapia.
En 3 análisis retrospectivos, que incluyeron un análisis conjunto de 5225 pacientes de 13 ensayos clínicos aleatorizados, se identificó un grupo de riesgo alto que tenía una tasa de SG del 50 % a los 5 años cuando las recaídas se presentaron dentro de los 24 meses posteriores a la quimioinmunoterapia de inducción. En un cuarto análisis retrospectivo de 296 pacientes que recibieron bendamustina y rituximab, se encontró una tasa de SG a 2 años del 38 % (intervalo de confianza [IC] 95 %, 20−55 %) en aquellos con progresión de la enfermedad antes de 24 meses (POD24). La mayoría de estos pacientes (76 %) presentaron transformación de la enfermedad (progresión histológica a linfoma difuso de células B grandes [LDCBG]). Estos pacientes con enfermedad POD24 de riesgo más alto representan una población de referencia para los ensayos clínicos.
No hay diferencias reproducibles en la supervivencia sin enfermedad ni en la SG del linfoma folicular de células hendidas pequeñas y el linfoma folicular mixto de células hendidas pequeñas y grandes.
Abordajes terapéuticos
A menudo el linfoma folicular tiene una evolución clínica de escasa malignidad y algunos pacientes son asintomáticos, por lo que la observación cautelosa continúa siendo el estándar de atención después de la primera consulta y para los pacientes con recaída asintomática de crecimiento lento. Cuando se requiere terapia, es posible usar numerosas opciones en secuencias diferentes con resultados equivalentes de la SG al cabo de 5 a 10 años. El rituximab se administra solo o en combinación con varias opciones de quimioterapia. Este fármaco también se puede combinar con el inmunomodulador lenalidomida para evitar la toxicidad a corto y largo plazo de los citotóxicos. Otro anticuerpo monoclonal anti-CD20, el obinutuzumab, a veces se administra con quimioterapia combinada. Los inhibidores de la fosfatidilinositol-3–cinasa (PI3K) también son eficaces en pacientes con enfermedad en recaída o resistente al tratamiento. Las células T con receptor de antígeno quimérico (CAR) dirigido a CD19 en ocasiones se usan en pacientes con progresión de la enfermedad después de dos o más líneas de tratamiento anteriores. En este entorno también es posible usar mosunetuzumab, un captador biespecífico de células T CD3 dirigido a CD20. Se puede considerar la terapia de consolidación para la enfermedad en recaída tras la terapia de reinducción con trasplante de células madre (TCM) autógeno o alogénico.
El linfoma folicular in situ y el linfoma folicular primario de duodeno son variantes de crecimiento lento que con poca frecuencia progresan o requieren tratamiento. El linfoma folicular ganglionar de tipo pediátrico tiene un comportamiento de crecimiento lento y rara vez recidiva; los pacientes adultos con esta variante histológica se caracterizan por una falta de reordenamiento de BCL2 junto con un índice de proliferación de Ki-67 superior al 30 % y una presentación localizada en estadio I.
Los pacientes con linfoma de crecimiento lento a veces recaen y presentan un tipo histológico más maligno o agresivo. Si el modelo clínico de la recaída indica que la enfermedad es más maligna, se deberá tomar una biopsia si esta es viable. Si se confirma que la enfermedad se convirtió a un tipo histológico más agresivo, se debe cambiar el tratamiento a un régimen que corresponda al tipo histológico. El crecimiento rápido o el crecimiento discordante entre varios sitios de la enfermedad en ocasiones son indicadores de una conversión histológica. En una revisión retrospectiva de 325 pacientes diagnosticados entre 1972 y 1999, el riesgo de transformación histológica fue del 30 % a los 10 años. En esta serie, los factores de riesgo alto para la transformación histológica posterior fueron el estadio avanzado, el FLIPI de riesgo alto y la conducta expectante (en lugar de iniciar el tratamiento en el momento del diagnóstico). La tasa de SG a 5 años fue de más del 50 % para los pacientes cuya enfermedad presentó una transformación histológica maligna comprobada mediante biopsia en varios estudios multicéntricos de cohortes en los que se usó rituximab con quimioterapia a base de antraciclina o derivados del platino, o un tratamiento similar seguido de TCM alogénico o autógeno.
En un estudio prospectivo no aleatorizado, tras una mediana de seguimiento de 6,8 años, 379 (14 %) pacientes de 2652 presentaron transformación posterior de la enfermedad a un tipo histológico más maligno después de un diagnóstico inicial de linfoma folicular.[Nivel de evidencia C3] La mediana de SG tras la transformación posterior fue de 5 años; sin embargo, entre 47 pacientes con evidencia de transformación y linfoma folicular en el momento del diagnóstico inicial, la SG no fue más precaria que la de los otros pacientes que no presentaron transformación (tasa de SG a 5 años, 88 %; IC 95 %, 74–95 %).
El tratamiento del linfoma folicular de grado 3b es similar al del LDCBG. Para obtener más información, consultar la sección Linfoma no Hodgkin de células B de crecimiento rápido.
Linfoma linfoplasmocítico (macroglobulinemia de Waldenström)
El linfoma linfoplasmocítico, también llamado linfoma linfoplasmocitario, linfoplasmácitico o macroglobulinemia de Waldenström, por lo general se relaciona con una paraproteína monoclonal sérica de tipo inmunoglobulina M (IgM). La mayoría de los pacientes presentan compromiso medular, ganglionar y esplénico, y en algunos quizás se observe síndrome de hiperviscosidad. La mayoría de los pacientes con macroglobulinemia de Waldenström son portadores de una mutación en MYD88 lo que algunos patólogos consideran indicador de la enfermedad. Es posible que otros linfomas también se relacionen con paraproteínas séricas. Los pacientes con linfoma linfoplasmocítico se deben examinar para detectar infección por el virus de la hepatitis C asociada.
En ocasiones se vigila a los pacientes asintomáticos para identificar signos de progresión de la enfermedad, sin necesidad de administrar quimioterapia inmediata .
Los factores pronósticos relacionados con síntomas que exigen tratamiento son los siguientes:
- Edad de 70 años o mayor.
- Microglobulina β2 de 3 mg/dl o más.
- Aumento de las concentraciones séricas de LDH.
Abordajes terapéuticos
El tratamiento del linfoma linfoplasmocítico es similar al de otros linfomas de grado bajo, en especial al tratamiento del linfoma linfocítico difuso de células pequeñas o de la leucemia linfocítica crónica. Si la viscosidad sérica relativa con respecto al agua es mayor de 4, el paciente quizás tenga síntomas de hiperviscosidad. La plasmaféresis es útil para tratar los síntomas agudos transitorios, como la retinopatía, la insuficiencia cardíaca congestiva y la disfunción del sistema nervioso central (SNC), pero en ocasiones se combina con quimioterapia para lograr un control prolongado de la enfermedad. Los pacientes sintomáticos con una viscosidad sérica de 4 o más baja se suelen tratar con quimioinmunoterapia o terapias biológicas dirigidas. Es posible que se necesite administrar tratamiento a los pacientes con enfermedad por crioaglutininas crónica para corregir anemia hemolítica; a menudo se emplea rituximab, bendamustina y corticoesteroides. En ocasiones, se necesita una habitación climatizada para los pacientes cuyas crioaglutininas se activan incluso ante el menor escalofrío. Es posible que sutimlimab, un anticuerpo monoclonal de tipo inmunoglobulina G4 que inhibe de manera selectiva la vía del complemento en C15, reduzca la hemólisis cuando las terapias dirigidas al linfoma linfoplasmocítico resultan ineficaces.
Los regímenes de primera línea incluyen zanubrutinib (inhibidor de la tirosina–cinasa de Bruton [BTK]), rituximab e ibrutinib (otro inhibidor de la BTK), rituximab solo, análogos nucleosídicos, y alquilantes, en monoterapia o quimioterapia combinada. En un ensayo prospectivo aleatorizado, 150 pacientes asintomáticos (incluso pacientes no tratados o con enfermedad recidivante) recibieron ibrutinib con rituximab o rituximab con un placebo. Después de una mediana de seguimiento de 50 meses, la tasa de SSP a 4,5 años favoreció al grupo de ibrutinib y rituximab (68 %; IC 95 %, 55–78 %) versus el grupo de rituximab y placebo (25 %; IC 95 %, 15–37 %) (cociente de riesgos instantáneos [CRI], 0,25; IC 95 %, 0,15–0,42; P < 0,0001). No hubo diferencias en la tasa de SG a 30 meses entre los 2 grupos (92–94 %).[Nivel de evidencia B1] El zanubrutinib se comparó con el ibrutinib en un ensayo clínico prospectivo aleatorizado de 164 pacientes con recaída de la enfermedad y 38 pacientes que no habían sido tratados. Al cabo de una mediana de seguimiento de 44,4 meses, la tasa de SSP fue similar en los 2 grupos, del 70 % al 78 % (CRI, 0,63; IC 95 %, 0,36–1,12) y la tasa de SG fue la misma en los dos grupos, del 85 % al 87 % (CRI, 0,75; IC 95 %, 0,36–1,59). El grupo de zanubrutinib presentó menos casos de fibrilación auricular (11 vs. 1) y un 50 % menos casos de hipertensión (no se notificaron los datos estadísticos).[Nivel de evidencia C3] La inhibición de la BTK con ibrutinib permitió que los 13 pacientes con anemia hemolítica autoinmunitaria mediada por anticuerpos fríos y que tenían acrocianosis lograran la remisión clínica con independencia del tipo patológico subyacente o del estado de MYD88.[Nivel de evidencia C3]
Los pacientes sin tratamiento previo que recibieron rituximab tuvieron tasas de respuesta del 60 % al 80 %, pero se necesita vigilar de cerca la IgM sérica por un posible aumento repentino de esta paraproteína al comienzo del tratamiento.[Nivel de evidencia C3] El aumento de la IgM al iniciar el rituximab se evita con el uso simultáneo de un alquilante, como ciclofosfamida o los inhibidores del proteosoma, bortezomib o ixazomib. Una combinación de bortezomib, dexametasona y rituximab se ha usado para evitar que la IgM vuelva a aumentar. Los pacientes con linfoma linfoplasmocítico que recibieron los análogos de nucleósidos cladribina y fludarabina, sin tratamiento previo, mostraron tasas de respuesta similares.[Nivel de evidencia C3] También obtienen tasas de respuesta similares los pacientes que recibieron monoterapias de alquilantes, como la bendamustina, el bortezomib o el venetoclax, y una quimioterapia combinada con rituximab o sin este.[Nivel de evidencia C3] En el caso poco frecuente de linfoma linfoplasmocítico que afecta el SNC (síndrome de Bing-Neel), el ibrutinib produjo una tasa de respuesta del 85 % en una serie anecdótica de 28 pacientes.[Nivel de evidencia C3]
La terapia mielosupresora con soporte de células madre hematopoyéticas autógenas o alogénicas está en evaluación clínica. Los candidatos a este abordaje deben evitar el uso a largo plazo de alquilantes o análogos de nucleósidos de purinas, que pueden agotar las células madre hematopoyéticas o predisponer a los pacientes a mielodisplasia o leucemia aguda. Después de una recaída durante la terapia con alquilantes, 92 pacientes con linfoma linfoplasmocítico se asignaron de manera aleatoria para recibir fludarabina o ciclofosfamida, doxorrubicina y prednisona. Aunque la supervivencia sin recaída favoreció la fludarabina (mediana de duración de 19 vs. 3 meses, P < 0,01), no se observaron diferencias en la SG.[Nivel de evidencia B1]
Linfoma de zona marginal
Cuando los linfomas de zona marginal comprometen los ganglios, se llaman linfomas de células B monocitoides o linfomas ganglionares de células B de zona marginal. Cuando comprometen sitios extraganglionares (por ejemplo, tubo digestivo, tiroides, pulmón, mama, órbita y piel), se llaman linfomas de tejido linfoide asociado a mucosas (MALT). El linfoma esplénico de zona marginal es una entidad clínica distinta que, por lo general, se presenta con esplenomegalia masiva. Una variante del linfoma MALT se conoce como enfermedad inmunoproliferativa del intestino delgado (EIPID). Un índice pronóstico de todos los linfomas de zona marginal incluye 3 factores de pronóstico adverso: edad de 70 años o más, enfermedad en estadio III o estadio IV, y concentración elevada de LDH. Menos del 10 % de los pacientes presentan una transformación a un linfoma de grado más alto. En una revisión retrospectiva, los factores de riesgo de transformación incluyeron la concentración elevada de LDH, más de 4 sitios ganglionares en el momento del diagnóstico inicial de linfoma de zona marginal y el fracaso en lograr una respuesta completa después del tratamiento inicial.
Tejido linfoide asociado a mucosa gástrica
Muchos pacientes tienen antecedentes de una enfermedad autoinmunitaria, como la tiroiditis de Hashimoto, el síndrome de Sjögren o una gastritis por Helicobacter pylori. La mayoría de los pacientes presentan una enfermedad extraganglionar en estadio I o estadio II, a menudo en el estómago. La mayoría de los casos de compromiso gástrico localizado quizás se resuelvan con el tratamiento de la infección por Helicobacter pylori. Después de administrar regímenes estándar con antibióticos, se observó en la endoscopia resolución del tejido linfoide asociado a mucosa (MALT) gástrica en 50 % de los pacientes al cabo de 3 meses. Es posible que otros pacientes se curen después de 12 a 18 meses de observación. Tras una mediana de seguimiento de 5 años, entre los pacientes que logran una remisión completa, 30 % tienen monoclonalidad asociada con un reordenamiento de la cadena pesada de las inmunoglobulinas detectada en biopsias de estómago. Se desconoce la implicación clínica de este hallazgo. La translocación t(11;18) en pacientes con MALT gástrica predice una respuesta precaria al tratamiento antibiótico y a la quimioterapia con alquilantes orales, además de pruebas negativas para la infección por H. pylori. Se ha hecho el seguimiento exitoso, de los pacientes con enfermedad asintomática estable y biopsias positivas persistentes, con un abordaje de observación cautelosa hasta el momento que progresa la enfermedad. Los pacientes con progresión de la enfermedad se tratan con radioterapia, rituximab, cirugía (gastrectomía total o gastrectomía parcial y radioterapia), quimioterapia, o terapia de modalidad combinada. La ecografía endoscópica quizás ayude a los profesionales clínicos a vigilar la respuesta de estos pacientes. En 4 series de casos que incluyeron a más de 100 pacientes con LDCBG en estadio IE o IIE con MALT o sin este (positivo para H. pylori), se notificaron remisiones completas duraderas en más del 50 % de los pacientes tras el tratamiento del H. pylori.
Tejido linfoide asociado a mucosa extragástrica
El compromiso localizado de otros sitios se puede tratar con radioterapia o cirugía. En algunas series, los pacientes con linfoma MALT extragástrico tienen una tasa de recaída más alta que los pacientes con linfoma MALT gástrico, y presentan recaídas después de muchos años, incluso décadas. Muchas de estas recidivas comprometen sitios de MALT diferentes a la ubicación original. Cuando se disemina a los ganglios linfáticos, la médula ósea o la sangre, esta entidad se comporta como otros linfomas de grado bajo. En un ensayo prospectivo aleatorizado de 401 pacientes con MALT extragástrico y extraganglionar se comparó el clorambucilo solo versus rituximab y clorambucilo versus rituximab solo. Después de una mediana de seguimiento de 7,4 años, la supervivencia sin complicaciones fue del 68 % en el grupo de rituximab y clorambucilo, del 51 % en el grupo de rituximab solo y del 50 % en el grupo de clorambucilo solo (P = 0,0009). Sin embargo, la tasa de SG a 5 años fue del 90 % en todos los grupos. En una revisión de la bibliografía que incluyó a 131 pacientes, en los pacientes con MALT de anejos oculares, el tratamiento con antibióticos dirigidos a Chlamydia psittaci produjo remisiones duraderas en casi la mitad de los pacientes.[Nivel de evidencia C3] Estas respuestas a la doxiciclina se observan sobre todo en ensayos italianos y con menor frecuencia en ensayos realizados en otros lugares geográficos. Los linfomas de células B grandes de sitios MALT se clasifican y tratan como linfomas difusos de células grandes. En una revisión retrospectiva grande de MALT de anejos oculares primario, se encontró que después de 10 años de seguimiento, en el 4 % de los pacientes en estadio I tratados con radioterapia se presentó transformación a LDCBG y el 3 % de ellos presentaron compromiso del SNC.
Linfoma ganglionar de zona marginal
Los pacientes con linfoma ganglionar de zona marginal (linfoma de células B monocitoides) se tratan con observación cautelosa o con terapias como se describe para el linfoma linfoplasmocítico. Se puede usar rituximab solo o combinado con citotóxicos (como bendamustina). El zanubrutinib está aprobado para los pacientes con recaída de la enfermedad después de recibir un régimen que contiene rituximab. Esta aprobación se basa en un estudio de fase II de un solo grupo. Después de una mediana de seguimiento de 15,7 meses, la tasa de respuesta general fue del 68,2 % y la tasa de respuesta completa fue del 25,8 %. La mediana de duración de la respuesta fue del 93 % a los 12 meses.[Nivel de evidencia C3] El ibrutinib también mostró una eficacia similar en pacientes con linfoma de zona marginal en recaída.[Nivel de evidencia C3] Al igual que con el linfoma folicular, los pacientes con POD24 que necesitaron iniciar el tratamiento tuvieron un pronóstico más precario (tasa de SG a 3 años del 53 %) que los pacientes sin POD24 (tasa de SG a 3 años del 95 %). Entre los pacientes con infección concomitante por el virus de la hepatitis C (VHC), del 40 % al 60 % lograron una remisión completa o parcial después de la pérdida del RNA del VHC detectable con tratamiento antivírico.[Nivel de evidencia C3]
Linfoma abdominal mediterráneo
La enfermedad conocida por varios nombres, como el linfoma abdominal mediterráneo, la enfermedad de cadena pesada o la EIPID, que se presenta en adultos jóvenes en los países del Mediterráneo oriental, es otra versión del linfoma MALT, que responde a los antibióticos en sus estadios iniciales. Se identificó que Campylobacter jejuni es una de las especies bacterianas relacionadas con el EIPID, y que el tratamiento antibiótico quizás produzca remisión de la enfermedad.
Linfoma esplénico de zona marginal
El linfoma esplénico de zona marginal es un linfoma de crecimiento lento que se caracteriza por esplenomegalia masiva y compromiso de la sangre periférica y la médula ósea, por lo general sin adenopatía. Este tipo de linfoma también se conoce como linfoma esplénico con linfocitos vellosos. La esplenectomía a veces produce remisión prolongada.
El tratamiento es similar al de otros linfomas de grado bajo y por lo general incluye rituximab solo o rituximab en combinación con análogos de las purinas o quimioterapia con alquilantes. El linfoma esplénico de zona marginal no responde tan bien a la quimioterapia que suele ser eficaz para la leucemia linfocítica crónica. Entre un pequeño número de pacientes con linfoma esplénico de zona marginal (linfoma esplénico con linfocitos vellosos) e infección por el VHC, la mayoría lograron una remisión completa o parcial tras la pérdida del RNA del VHC mediante el tratamiento con interferón-α, con ribavirina o sin esta.; [Nivel de evidencia C3] En contraste, no se observaron respuestas al interferón en 6 pacientes sin infección por el VHC.
References
- Armitage JO, Weisenburger DD: New approach to classifying non-Hodgkin's lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin's Lymphoma Classification Project. J Clin Oncol 16 (8): 2780-95, 1998.
- A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin's lymphoma. The Non-Hodgkin's Lymphoma Classification Project. Blood 89 (11): 3909-18, 1997.
- Society for Hematopathology Program: Society for Hematopathology Program. Am J Surg Pathol 21 (1): 114-121, 1997.
- López-Guillermo A, Cabanillas F, McDonnell TI, et al.: Correlation of bcl-2 rearrangement with clinical characteristics and outcome in indolent follicular lymphoma. Blood 93 (9): 3081-7, 1999.
- Peterson BA, Petroni GR, Frizzera G, et al.: Prolonged single-agent versus combination chemotherapy in indolent follicular lymphomas: a study of the cancer and leukemia group B. J Clin Oncol 21 (1): 5-15, 2003.
- Swenson WT, Wooldridge JE, Lynch CF, et al.: Improved survival of follicular lymphoma patients in the United States. J Clin Oncol 23 (22): 5019-26, 2005.
- Liu Q, Fayad L, Cabanillas F, et al.: Improvement of overall and failure-free survival in stage IV follicular lymphoma: 25 years of treatment experience at The University of Texas M.D. Anderson Cancer Center. J Clin Oncol 24 (10): 1582-9, 2006.
- Kahl BS, Yang DT: Follicular lymphoma: evolving therapeutic strategies. Blood 127 (17): 2055-63, 2016.
- Ardeshna KM, Smith P, Norton A, et al.: Long-term effect of a watch and wait policy versus immediate systemic treatment for asymptomatic advanced-stage non-Hodgkin lymphoma: a randomised controlled trial. Lancet 362 (9383): 516-22, 2003.
- Armitage JO, Longo DL: Is watch and wait still acceptable for patients with low-grade follicular lymphoma? Blood 127 (23): 2804-8, 2016.
- Solal-Céligny P, Roy P, Colombat P, et al.: Follicular lymphoma international prognostic index. Blood 104 (5): 1258-65, 2004.
- Perea G, Altés A, Montoto S, et al.: Prognostic indexes in follicular lymphoma: a comparison of different prognostic systems. Ann Oncol 16 (9): 1508-13, 2005.
- Buske C, Hoster E, Dreyling M, et al.: The Follicular Lymphoma International Prognostic Index (FLIPI) separates high-risk from intermediate- or low-risk patients with advanced-stage follicular lymphoma treated front-line with rituximab and the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) with respect to treatment outcome. Blood 108 (5): 1504-8, 2006.
- Federico M, Bellei M, Marcheselli L, et al.: Follicular lymphoma international prognostic index 2: a new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J Clin Oncol 27 (27): 4555-62, 2009.
- Bachy E, Maurer MJ, Habermann TM, et al.: A simplified scoring system in de novo follicular lymphoma treated initially with immunochemotherapy. Blood 132 (1): 49-58, 2018.
- Mir F, Mattiello F, Grigg A, et al.: Follicular Lymphoma Evaluation Index (FLEX): A new clinical prognostic model that is superior to existing risk scores for predicting progression-free survival and early treatment failure after frontline immunochemotherapy. Am J Hematol 95 (12): 1503-1510, 2020.
- Casulo C, Byrtek M, Dawson KL, et al.: Early Relapse of Follicular Lymphoma After Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone Defines Patients at High Risk for Death: An Analysis From the National LymphoCare Study. J Clin Oncol 33 (23): 2516-22, 2015.
- Shi Q, Flowers CR, Hiddemann W, et al.: Thirty-Month Complete Response as a Surrogate End Point in First-Line Follicular Lymphoma Therapy: An Individual Patient-Level Analysis of Multiple Randomized Trials. J Clin Oncol 35 (5): 552-560, 2017.
- Casulo C, Dixon JG, Le-Rademacher J, et al.: Validation of POD24 as a robust early clinical end point of poor survival in FL from 5225 patients on 13 clinical trials. Blood 139 (11): 1684-1693, 2022.
- Freeman CL, Kridel R, Moccia AA, et al.: Early progression after bendamustine-rituximab is associated with high risk of transformation in advanced stage follicular lymphoma. Blood 134 (9): 761-764, 2019.
- Brice P, Bastion Y, Lepage E, et al.: Comparison in low-tumor-burden follicular lymphomas between an initial no-treatment policy, prednimustine, or interferon alfa: a randomized study from the Groupe d'Etude des Lymphomes Folliculaires. Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 15 (3): 1110-7, 1997.
- Young RC, Longo DL, Glatstein E, et al.: The treatment of indolent lymphomas: watchful waiting v aggressive combined modality treatment. Semin Hematol 25 (2 Suppl 2): 11-6, 1988.
- Luminari S, Ferrari A, Manni M, et al.: Long-Term Results of the FOLL05 Trial Comparing R-CVP Versus R-CHOP Versus R-FM for the Initial Treatment of Patients With Advanced-Stage Symptomatic Follicular Lymphoma. J Clin Oncol 36 (7): 689-696, 2018.
- Lockmer S, Østenstad B, Hagberg H, et al.: Chemotherapy-Free Initial Treatment of Advanced Indolent Lymphoma Has Durable Effect With Low Toxicity: Results From Two Nordic Lymphoma Group Trials With More Than 10 Years of Follow-Up. J Clin Oncol : JCO1800262, 2018.
- Cartron G, Bachy E, Tilly H, et al.: Randomized Phase III Trial Evaluating Subcutaneous Rituximab for the First-Line Treatment of Low-Tumor Burden Follicular Lymphoma: Results of a LYSA Study. J Clin Oncol 41 (19): 3523-3533, 2023.
- Morschhauser F, Fowler NH, Feugier P, et al.: Rituximab plus Lenalidomide in Advanced Untreated Follicular Lymphoma. N Engl J Med 379 (10): 934-947, 2018.
- Leonard JP, Trnený M, Izutsu K, et al.: Augment: a phase III randomized study of lenalidomide Plus rituximab (R2) vs rituximab/placebo in patients with relapsed/refractory indolent non-Hodgkin lymphoma. [Abstract] Blood 132 (Suppl 1): A-445, 2018.
- Zucca E, Rondeau S, Vanazzi A, et al.: Short regimen of rituximab plus lenalidomide in follicular lymphoma patients in need of first-line therapy. Blood 134 (4): 353-362, 2019.
- Marcus R, Davies A, Ando K, et al.: Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N Engl J Med 377 (14): 1331-1344, 2017.
- Dreyling M, Santoro A, Mollica L, et al.: Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol 35 (35): 3898-3905, 2017.
- Jacobson CA, Chavez JC, Sehgal AR, et al.: Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol 23 (1): 91-103, 2022.
- Bartlett NL, Sehn LH, Matasar MJ, et al.: Mosunetuzumab monotherapy demonstrates durable efficacy with a manageable safety profile in patients with relapsed/refractory follicular lymphoma who received ≥2 prior therapies: updated results from a pivotal phase II study. [Abstract] Blood 140 (Suppl 1): A-610, 1467-70, 2022.
- Schaaf M, Reiser M, Borchmann P, et al.: High-dose therapy with autologous stem cell transplantation versus chemotherapy or immuno-chemotherapy for follicular lymphoma in adults. Cochrane Database Syst Rev 1: CD007678, 2012.
- Schmatz AI, Streubel B, Kretschmer-Chott E, et al.: Primary follicular lymphoma of the duodenum is a distinct mucosal/submucosal variant of follicular lymphoma: a retrospective study of 63 cases. J Clin Oncol 29 (11): 1445-51, 2011.
- Jegalian AG, Eberle FC, Pack SD, et al.: Follicular lymphoma in situ: clinical implications and comparisons with partial involvement by follicular lymphoma. Blood 118 (11): 2976-84, 2011.
- Louissaint A, Ackerman AM, Dias-Santagata D, et al.: Pediatric-type nodal follicular lymphoma: an indolent clonal proliferation in children and adults with high proliferation index and no BCL2 rearrangement. Blood 120 (12): 2395-404, 2012.
- Sarkozy C, Trneny M, Xerri L, et al.: Risk Factors and Outcomes for Patients With Follicular Lymphoma Who Had Histologic Transformation After Response to First-Line Immunochemotherapy in the PRIMA Trial. J Clin Oncol 34 (22): 2575-82, 2016.
- Tsimberidou AM, O'Brien S, Khouri I, et al.: Clinical outcomes and prognostic factors in patients with Richter's syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J Clin Oncol 24 (15): 2343-51, 2006.
- Montoto S, Davies AJ, Matthews J, et al.: Risk and clinical implications of transformation of follicular lymphoma to diffuse large B-cell lymphoma. J Clin Oncol 25 (17): 2426-33, 2007.
- Villa D, Crump M, Panzarella T, et al.: Autologous and allogeneic stem-cell transplantation for transformed follicular lymphoma: a report of the Canadian blood and marrow transplant group. J Clin Oncol 31 (9): 1164-71, 2013.
- Williams CD, Harrison CN, Lister TA, et al.: High-dose therapy and autologous stem-cell support for chemosensitive transformed low-grade follicular non-Hodgkin's lymphoma: a case-matched study from the European Bone Marrow Transplant Registry. J Clin Oncol 19 (3): 727-35, 2001.
- Wagner-Johnston ND, Link BK, Byrtek M, et al.: Outcomes of transformed follicular lymphoma in the modern era: a report from the National LymphoCare Study (NLCS). Blood 126 (7): 851-7, 2015.
- Leblond V, Kastritis E, Advani R, et al.: Treatment recommendations from the Eighth International Workshop on Waldenström's Macroglobulinemia. Blood 128 (10): 1321-8, 2016.
- Treon SP, Xu L, Yang G, et al.: MYD88 L265P somatic mutation in Waldenström's macroglobulinemia. N Engl J Med 367 (9): 826-33, 2012.
- Dhodapkar MV, Hoering A, Gertz MA, et al.: Long-term survival in Waldenstrom macroglobulinemia: 10-year follow-up of Southwest Oncology Group-directed intergroup trial S9003. Blood 113 (4): 793-6, 2009.
- Ansell SM, Kyle RA, Reeder CB, et al.: Diagnosis and management of Waldenström macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines. Mayo Clin Proc 85 (9): 824-33, 2010.
- Kapoor P, Ansell SM, Fonseca R, et al.: Diagnosis and Management of Waldenström Macroglobulinemia: Mayo Stratification of Macroglobulinemia and Risk-Adapted Therapy (mSMART) Guidelines 2016. JAMA Oncol 3 (9): 1257-1265, 2017.
- Dimopoulos MA, Kastritis E: How I treat Waldenström macroglobulinemia. Blood 134 (23): 2022-2035, 2019.
- Gertz MA: Waldenstrom Macroglobulinemia: Tailoring Therapy for the Individual. J Clin Oncol 40 (23): 2600-2608, 2022.
- Röth A, Berentsen S, Barcellini W, et al.: Sutimlimab in patients with cold agglutinin disease: results of the randomized placebo-controlled phase 3 CADENZA trial. Blood 140 (9): 980-991, 2022.
- Buske C, Dimopoulos MA, Grunenberg A, et al.: Bortezomib-Dexamethasone, Rituximab, and Cyclophosphamide as First-Line Treatment for Waldenström's Macroglobulinemia: A Prospectively Randomized Trial of the European Consortium for Waldenström's Macroglobulinemia. J Clin Oncol 41 (14): 2607-2616, 2023.
- Leblond V, Johnson S, Chevret S, et al.: Results of a randomized trial of chlorambucil versus fludarabine for patients with untreated Waldenström macroglobulinemia, marginal zone lymphoma, or lymphoplasmacytic lymphoma. J Clin Oncol 31 (3): 301-7, 2013.
- Buske C, Tedeschi A, Trotman J, et al.: Ibrutinib Plus Rituximab Versus Placebo Plus Rituximab for Waldenström's Macroglobulinemia: Final Analysis From the Randomized Phase III iNNOVATE Study. J Clin Oncol 40 (1): 52-62, 2022.
- Tam CS, Opat S, D'Sa S, et al.: A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood 136 (18): 2038-2050, 2020.
- Dimopoulos MA, Oriol A, Nahi H, et al.: Overall Survival With Daratumumab, Lenalidomide, and Dexamethasone in Previously Treated Multiple Myeloma (POLLUX): A Randomized, Open-Label, Phase III Trial. J Clin Oncol 41 (8): 1590-1599, 2023.
- Jalink M, Berentsen S, Castillo JJ, et al.: Effect of ibrutinib treatment on hemolytic anemia and acrocyanosis in cold agglutinin disease/cold agglutinin syndrome. Blood 138 (20): 2002-2005, 2021.
- Gertz MA, Anagnostopoulos A, Anderson K, et al.: Treatment recommendations in Waldenstrom's macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom's Macroglobulinemia. Semin Oncol 30 (2): 121-6, 2003.
- Dimopoulos MA, Zervas C, Zomas A, et al.: Treatment of Waldenström's macroglobulinemia with rituximab. J Clin Oncol 20 (9): 2327-33, 2002.
- Treon SP, Branagan AR, Hunter Z, et al.: Paradoxical increases in serum IgM and viscosity levels following rituximab in Waldenstrom's macroglobulinemia. Ann Oncol 15 (10): 1481-3, 2004.
- Dimopoulos MA, Chen C, Kastritis E, et al.: Bortezomib as a treatment option in patients with Waldenström macroglobulinemia. Clin Lymphoma Myeloma Leuk 10 (2): 110-7, 2010.
- Gavriatopoulou M, García-Sanz R, Kastritis E, et al.: BDR in newly diagnosed patients with WM: final analysis of a phase 2 study after a minimum follow-up of 6 years. Blood 129 (4): 456-459, 2017.
- Kersten MJ, Amaador K, Minnema MC, et al.: Combining Ixazomib With Subcutaneous Rituximab and Dexamethasone in Relapsed or Refractory Waldenström's Macroglobulinemia: Final Analysis of the Phase I/II HOVON124/ECWM-R2 Study. J Clin Oncol 40 (1): 40-51, 2022.
- Treon SP, Ioakimidis L, Soumerai JD, et al.: Primary therapy of Waldenström macroglobulinemia with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial 05-180. J Clin Oncol 27 (23): 3830-5, 2009.
- Dimopoulos MA, García-Sanz R, Gavriatopoulou M, et al.: Primary therapy of Waldenstrom macroglobulinemia (WM) with weekly bortezomib, low-dose dexamethasone, and rituximab (BDR): long-term results of a phase 2 study of the European Myeloma Network (EMN). Blood 122 (19): 3276-82, 2013.
- Treon SP, Tripsas CK, Meid K, et al.: Carfilzomib, rituximab, and dexamethasone (CaRD) treatment offers a neuropathy-sparing approach for treating Waldenström's macroglobulinemia. Blood 124 (4): 503-10, 2014.
- Dimopoulos MA, Alexanian R: Waldenstrom's macroglobulinemia. Blood 83 (6): 1452-9, 1994.
- Laszlo D, Andreola G, Rigacci L, et al.: Rituximab and subcutaneous 2-chloro-2'-deoxyadenosine combination treatment for patients with Waldenstrom macroglobulinemia: clinical and biologic results of a phase II multicenter study. J Clin Oncol 28 (13): 2233-8, 2010.
- García-Sanz R, Montoto S, Torrequebrada A, et al.: Waldenström macroglobulinaemia: presenting features and outcome in a series with 217 cases. Br J Haematol 115 (3): 575-82, 2001.
- Buske C, Hoster E, Dreyling M, et al.: The addition of rituximab to front-line therapy with CHOP (R-CHOP) results in a higher response rate and longer time to treatment failure in patients with lymphoplasmacytic lymphoma: results of a randomized trial of the German Low-Grade Lymphoma Study Group (GLSG). Leukemia 23 (1): 153-61, 2009.
- Ghobrial IM, Hong F, Padmanabhan S, et al.: Phase II trial of weekly bortezomib in combination with rituximab in relapsed or relapsed and refractory Waldenstrom macroglobulinemia. J Clin Oncol 28 (8): 1422-8, 2010.
- Rummel MJ, Niederle N, Maschmeyer G, et al.: Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 381 (9873): 1203-10, 2013.
- Castillo JJ, Allan JN, Siddiqi T, et al.: Venetoclax in Previously Treated Waldenström Macroglobulinemia. J Clin Oncol 40 (1): 63-71, 2022.
- Castillo JJ, Itchaki G, Paludo J, et al.: Ibrutinib for the treatment of Bing-Neel syndrome: a multicenter study. Blood 133 (4): 299-305, 2019.
- Dreger P, Glass B, Kuse R, et al.: Myeloablative radiochemotherapy followed by reinfusion of purged autologous stem cells for Waldenström's macroglobulinaemia. Br J Haematol 106 (1): 115-8, 1999.
- Desikan R, Dhodapkar M, Siegel D, et al.: High-dose therapy with autologous haemopoietic stem cell support for Waldenström's macroglobulinaemia. Br J Haematol 105 (4): 993-6, 1999.
- Martin P, Chadburn A, Christos P, et al.: Intensive treatment strategies may not provide superior outcomes in mantle cell lymphoma: overall survival exceeding 7 years with standard therapies. Ann Oncol 19 (7): 1327-30, 2008.
- Kyriakou C, Canals C, Cornelissen JJ, et al.: Allogeneic stem-cell transplantation in patients with Waldenström macroglobulinemia: report from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 28 (33): 4926-34, 2010.
- Leleu X, Soumerai J, Roccaro A, et al.: Increased incidence of transformation and myelodysplasia/acute leukemia in patients with Waldenström macroglobulinemia treated with nucleoside analogs. J Clin Oncol 27 (2): 250-5, 2009.
- Leblond V, Lévy V, Maloisel F, et al.: Multicenter, randomized comparative trial of fludarabine and the combination of cyclophosphamide-doxorubicin-prednisone in 92 patients with Waldenström macroglobulinemia in first relapse or with primary refractory disease. Blood 98 (9): 2640-4, 2001.
- Zucca E, Bertoni F: The spectrum of MALT lymphoma at different sites: biological and therapeutic relevance. Blood 127 (17): 2082-92, 2016.
- Rossi D, Bertoni F, Zucca E: Marginal-Zone Lymphomas. N Engl J Med 386 (6): 568-581, 2022.
- Thieblemont C, Cascione L, Conconi A, et al.: A MALT lymphoma prognostic index. Blood 130 (12): 1409-1417, 2017.
- Alderuccio JP, Zhao W, Desai A, et al.: Risk Factors for Transformation to Higher-Grade Lymphoma and Its Impact on Survival in a Large Cohort of Patients With Marginal Zone Lymphoma From a Single Institution. J Clin Oncol : JCO1800138, 2018.
- Zullo A, Hassan C, Andriani A, et al.: Eradication therapy for Helicobacter pylori in patients with gastric MALT lymphoma: a pooled data analysis. Am J Gastroenterol 104 (8): 1932-7; quiz 1938, 2009.
- Nakamura S, Sugiyama T, Matsumoto T, et al.: Long-term clinical outcome of gastric MALT lymphoma after eradication of Helicobacter pylori: a multicentre cohort follow-up study of 420 patients in Japan. Gut 61 (4): 507-13, 2012.
- Wündisch T, Thiede C, Morgner A, et al.: Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol 23 (31): 8018-24, 2005.
- Ye H, Liu H, Raderer M, et al.: High incidence of t(11;18)(q21;q21) in Helicobacter pylori-negative gastric MALT lymphoma. Blood 101 (7): 2547-50, 2003.
- Lévy M, Copie-Bergman C, Gameiro C, et al.: Prognostic value of translocation t(11;18) in tumoral response of low-grade gastric lymphoma of mucosa-associated lymphoid tissue type to oral chemotherapy. J Clin Oncol 23 (22): 5061-6, 2005.
- Nakamura S, Ye H, Bacon CM, et al.: Clinical impact of genetic aberrations in gastric MALT lymphoma: a comprehensive analysis using interphase fluorescence in situ hybridisation. Gut 56 (10): 1358-63, 2007.
- Schechter NR, Yahalom J: Low-grade MALT lymphoma of the stomach: a review of treatment options. Int J Radiat Oncol Biol Phys 46 (5): 1093-103, 2000.
- Tsang RW, Gospodarowicz MK, Pintilie M, et al.: Stage I and II MALT lymphoma: results of treatment with radiotherapy. Int J Radiat Oncol Biol Phys 50 (5): 1258-64, 2001.
- Tsang RW, Gospodarowicz MK, Pintilie M, et al.: Localized mucosa-associated lymphoid tissue lymphoma treated with radiation therapy has excellent clinical outcome. J Clin Oncol 21 (22): 4157-64, 2003.
- Tsai HK, Li S, Ng AK, et al.: Role of radiation therapy in the treatment of stage I/II mucosa-associated lymphoid tissue lymphoma. Ann Oncol 18 (4): 672-8, 2007.
- De Leo AN, Bates JE, Lockney NA, et al.: Radiotherapy in Early-stage Gastric MALT: Improved Survival Without Increased Cardiac Death. Am J Clin Oncol 43 (11): 770-775, 2020.
- Martinelli G, Laszlo D, Ferreri AJ, et al.: Clinical activity of rituximab in gastric marginal zone non-Hodgkin's lymphoma resistant to or not eligible for anti-Helicobacter pylori therapy. J Clin Oncol 23 (9): 1979-83, 2005.
- Cogliatti SB, Schmid U, Schumacher U, et al.: Primary B-cell gastric lymphoma: a clinicopathological study of 145 patients. Gastroenterology 101 (5): 1159-70, 1991.
- Zinzani PL, Magagnoli M, Galieni P, et al.: Nongastrointestinal low-grade mucosa-associated lymphoid tissue lymphoma: analysis of 75 patients. J Clin Oncol 17 (4): 1254, 1999.
- Thieblemont C, Bastion Y, Berger F, et al.: Mucosa-associated lymphoid tissue gastrointestinal and nongastrointestinal lymphoma behavior: analysis of 108 patients. J Clin Oncol 15 (4): 1624-30, 1997.
- Pavlick AC, Gerdes H, Portlock CS: Endoscopic ultrasound in the evaluation of gastric small lymphocytic mucosa-associated lymphoid tumors. J Clin Oncol 15 (5): 1761-6, 1997.
- Morgner A, Miehlke S, Fischbach W, et al.: Complete remission of primary high-grade B-cell gastric lymphoma after cure of Helicobacter pylori infection. J Clin Oncol 19 (7): 2041-8, 2001.
- Chen LT, Lin JT, Shyu RY, et al.: Prospective study of Helicobacter pylori eradication therapy in stage I(E) high-grade mucosa-associated lymphoid tissue lymphoma of the stomach. J Clin Oncol 19 (22): 4245-51, 2001.
- Chen LT, Lin JT, Tai JJ, et al.: Long-term results of anti-Helicobacter pylori therapy in early-stage gastric high-grade transformed MALT lymphoma. J Natl Cancer Inst 97 (18): 1345-53, 2005.
- Kuo SH, Yeh KH, Wu MS, et al.: Helicobacter pylori eradication therapy is effective in the treatment of early-stage H pylori-positive gastric diffuse large B-cell lymphomas. Blood 119 (21): 4838-44; quiz 5057, 2012.
- Uno T, Isobe K, Shikama N, et al.: Radiotherapy for extranodal, marginal zone, B-cell lymphoma of mucosa-associated lymphoid tissue originating in the ocular adnexa: a multiinstitutional, retrospective review of 50 patients. Cancer 98 (4): 865-71, 2003.
- Bayraktar S, Bayraktar UD, Stefanovic A, et al.: Primary ocular adnexal mucosa-associated lymphoid tissue lymphoma (MALT): single institution experience in a large cohort of patients. Br J Haematol 152 (1): 72-80, 2011.
- Stefanovic A, Lossos IS: Extranodal marginal zone lymphoma of the ocular adnexa. Blood 114 (3): 501-10, 2009.
- Vazquez A, Khan MN, Sanghvi S, et al.: Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue of the salivary glands: a population-based study from 1994 to 2009. Head Neck 37 (1): 18-22, 2015.
- Raderer M, Streubel B, Woehrer S, et al.: High relapse rate in patients with MALT lymphoma warrants lifelong follow-up. Clin Cancer Res 11 (9): 3349-52, 2005.
- Sretenovic M, Colovic M, Jankovic G, et al.: More than a third of non-gastric malt lymphomas are disseminated at diagnosis: a single center survey. Eur J Haematol 82 (5): 373-80, 2009.
- Nathwani BN, Drachenberg MR, Hernandez AM, et al.: Nodal monocytoid B-cell lymphoma (nodal marginal-zone B-cell lymphoma). Semin Hematol 36 (2): 128-38, 1999.
- Raderer M, Wöhrer S, Streubel B, et al.: Assessment of disease dissemination in gastric compared with extragastric mucosa-associated lymphoid tissue lymphoma using extensive staging: a single-center experience. J Clin Oncol 24 (19): 3136-41, 2006.
- Zucca E, Conconi A, Martinelli G, et al.: Final Results of the IELSG-19 Randomized Trial of Mucosa-Associated Lymphoid Tissue Lymphoma: Improved Event-Free and Progression-Free Survival With Rituximab Plus Chlorambucil Versus Either Chlorambucil or Rituximab Monotherapy. J Clin Oncol 35 (17): 1905-1912, 2017.
- Kiesewetter B, Raderer M: Antibiotic therapy in nongastrointestinal MALT lymphoma: a review of the literature. Blood 122 (8): 1350-7, 2013.
- Grünberger B, Hauff W, Lukas J, et al.: 'Blind' antibiotic treatment targeting Chlamydia is not effective in patients with MALT lymphoma of the ocular adnexa. Ann Oncol 17 (3): 484-7, 2006.
- Kuo SH, Chen LT, Yeh KH, et al.: Nuclear expression of BCL10 or nuclear factor kappa B predicts Helicobacter pylori-independent status of early-stage, high-grade gastric mucosa-associated lymphoid tissue lymphomas. J Clin Oncol 22 (17): 3491-7, 2004.
- Desai A, Joag MG, Lekakis L, et al.: Long-term course of patients with primary ocular adnexal MALT lymphoma: a large single-institution cohort study. Blood 129 (3): 324-332, 2017.
- Opat S, Tedeschi A, Linton K, et al.: The MAGNOLIA Trial: Zanubrutinib, a Next-Generation Bruton Tyrosine Kinase Inhibitor, Demonstrates Safety and Efficacy in Relapsed/Refractory Marginal Zone Lymphoma. Clin Cancer Res 27 (23): 6323-6332, 2021.
- Noy A, de Vos S, Coleman M, et al.: Durable ibrutinib responses in relapsed/refractory marginal zone lymphoma: long-term follow-up and biomarker analysis. Blood Adv 4 (22): 5773-5784, 2020.
- Luminari S, Merli M, Rattotti S, et al.: Early progression as a predictor of survival in marginal zone lymphomas: an analysis from the FIL-NF10 study. Blood 134 (10): 798-801, 2019.
- Vallisa D, Bernuzzi P, Arcaini L, et al.: Role of anti-hepatitis C virus (HCV) treatment in HCV-related, low-grade, B-cell, non-Hodgkin's lymphoma: a multicenter Italian experience. J Clin Oncol 23 (3): 468-73, 2005.
- Merli M, Rattotti S, Spina M, et al.: Direct-Acting Antivirals as Primary Treatment for Hepatitis C Virus-Associated Indolent Non-Hodgkin Lymphomas: The BArT Study of the Fondazione Italiana Linfomi. J Clin Oncol 40 (35): 4060-4070, 2022.
- Isaacson PG: Gastrointestinal lymphoma. Hum Pathol 25 (10): 1020-9, 1994.
- Lecuit M, Abachin E, Martin A, et al.: Immunoproliferative small intestinal disease associated with Campylobacter jejuni. N Engl J Med 350 (3): 239-48, 2004.
- Arcaini L, Paulli M, Boveri E, et al.: Splenic and nodal marginal zone lymphomas are indolent disorders at high hepatitis C virus seroprevalence with distinct presenting features but similar morphologic and phenotypic profiles. Cancer 100 (1): 107-15, 2004.
- Arcaini L, Rossi D, Paulli M: Splenic marginal zone lymphoma: from genetics to management. Blood 127 (17): 2072-81, 2016.
- Bertoni F, Zucca E: State-of-the-art therapeutics: marginal-zone lymphoma. J Clin Oncol 23 (26): 6415-20, 2005.
- Parry-Jones N, Matutes E, Gruszka-Westwood AM, et al.: Prognostic features of splenic lymphoma with villous lymphocytes: a report on 129 patients. Br J Haematol 120 (5): 759-64, 2003.
- Arcaini L, Lazzarino M, Colombo N, et al.: Splenic marginal zone lymphoma: a prognostic model for clinical use. Blood 107 (12): 4643-9, 2006.
- Iannitto E, Ambrosetti A, Ammatuna E, et al.: Splenic marginal zone lymphoma with or without villous lymphocytes. Hematologic findings and outcomes in a series of 57 patients. Cancer 101 (9): 2050-7, 2004.
- Hermine O, Lefrère F, Bronowicki JP, et al.: Regression of splenic lymphoma with villous lymphocytes after treatment of hepatitis C virus infection. N Engl J Med 347 (2): 89-94, 2002.
- Kelaidi C, Rollot F, Park S, et al.: Response to antiviral treatment in hepatitis C virus-associated marginal zone lymphomas. Leukemia 18 (10): 1711-6, 2004.
Linfoma no Hodgkin de células B de crecimiento rápido
El linfoma no Hodgkin (LNH) de células B de crecimiento rápido (agresivo) incluye los siguientes subtipos:
- Linfoma difuso de células B grandes.
- Linfoma mediastínico primario de células B grandes.
- Linfoma intravascular de células B grandes (linfomatosis intravascular).
- Linfoma folicular (grado 3b).
- Linfoma de células de manto.
- Linfoma de Burkitt o linfoma difuso de células pequeñas no hendidas.
- Linfoma linfoblástico de células B.
- Linfoma primario de efusiones.
- Linfoma plasmoblástico (plasmablástico).
- Trastorno linfoproliferativo postrasplante polimórfico.
- Granulomatosis linfomatoide.
Linfoma difuso de células B grandes
El linfoma difuso de células B grandes (LDCBG) es el tipo más común de LNH y representa el 30 % de los casos recién diagnosticados. La mayoría de los pacientes presentan masas que crecen rápido, a menudo con síntomas locales y sistémicos (designados como síntomas B, con fiebre, hiperhidrosis nocturna recurrente o pérdida de peso). Para obtener más información sobre la pérdida de peso, consultar La nutrición en el tratamiento del cáncer.
Algunos casos de linfoma de células B grandes tienen un fondo de células T reactivas y a menudo histiocitos, en este caso se denomina linfoma de células T y de células B grandes rico en histiocitos. Este subtipo de linfoma de células grandes a menudo compromete el hígado, el bazo y la médula ósea; sin embargo, el desenlace es equivalente al de pacientes con LDCBG en estadios similares. En el momento del diagnóstico, algunos pacientes con LDCBG presentan de manera simultánea un componente de células B pequeñas de crecimiento lento. Si bien la supervivencia general (SG) es similar al del LDCBG de novo después de la quimioterapia multifarmacológica, hay un riesgo más alto de recaída de la enfermedad de crecimiento lento.
Pronóstico
La mayoría de los pacientes con enfermedad localizada se curan con una terapia de modalidad combinada o quimioterapia combinada sola. Entre los pacientes con enfermedad en estadio avanzado, el 50 % se curan con quimioterapia combinada a base de doxorrubicina y rituximab, por lo general, R-CHOP (rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona).
En el índice pronóstico internacional de la National Comprehensive Cancer Network (National Comprehensive Cancer Network International Prognostic Index [NCCN-IPI]) para el linfoma no Hodgkin (LNH) de crecimiento rápido (linfoma difuso de células grandes) se identifican los siguientes 5 factores de riesgo significativos para el pronóstico de la supervivencia general (SG) y los puntajes de riesgo relacionado:
- Edad.
- <40 años: 0.
- 41–60 años: 1.
- 61–75 años: 2.
- >75 años: 3.
- Estadio III o IV: 1.
- Estado funcional (EF) 2, 3 o 4: 1.
- Concentración sérica de lactato–deshidrogenasa (LDH).
- Normalizada: 0.
- >1x–3x: 1.
- >3x: 2.
- Número de sitios extraganglionares ≥2: 1.
Puntajes de riesgo:
- Bajo (0 o 1): Tasa de SG a 5 años, 96 %; tasa de supervivencia sin progresión (SSP), 91 %.
- Intermedio bajo (2 o 3): Tasa de SG a 5 años, 82 %; tasa de SSP, 74 %.
- Intermedio alto (4 o 5): Tasa de SG a 5 años, 64 %; tasa de SSP, 51 %.
- Alto (>6): Tasa de SG a 5 años, 33 %; tasa de SSP, 30 %.
Se utilizan modificaciones del IPI ajustadas por edad y estadio para los pacientes más jóvenes con enfermedad localizada. Los intervalos de tiempo más cortos entre el diagnóstico y el tratamiento son marcadores indirectos de factores biológicos de pronóstico precario.
El gen BCL2 y el reordenamiento del gen MYC o la sobreexpresión dual del gen MYC, o ambos, confieren un pronóstico particularmente precario. En este grupo de pacientes de riesgo alto se explora el uso de las terapias de dosis intensivas, las terapias de infusión, y la consolidación con trasplante de células madre (TCM). En una revisión retrospectiva, se evaluaron 159 pacientes de LDCBG sin tratamiento previo sometidos a pruebas genéticas para detectar mutación doble (double-hit) mediante hibridación fluorescente in situ (FISH), que lograron una respuesta completa. La terapia de inducción no alteró la supervivencia sin recaída a 3 años ni la SG cuando se usó un TCM autógeno.
En una revisión retrospectiva con 117 pacientes de LDCBG en recaída o resistente al tratamiento sometidos a TCM autógeno, se encontró que la tasa de SG a 4 años fue del 25 % para los linfomas con mutación doble (reordenamientos de BCL2 y MYC), del 61 % para los linfomas con expresión doble (sin reordenamientos, pero con aumento de la expresión de BCL2 y MYC) y del 70 % para los pacientes sin estas características. Los pacientes en riesgo alto de recaída se consideran aptos para participar en ensayos clínicos.
La obtención de perfiles moleculares de expresión génica mediante micromatrices de ADN quizás sirva en el futuro para ayudar a estratificar a los pacientes en grupos de tratamiento dirigido a dianas específicas y para mejorar la predicción de la supervivencia después de la quimioterapia estándar. Por ejemplo, los linfomas de células B grandes positivos para la cinasa del linfoma anaplásico (ALK) son muy poco frecuentes y no responden bien al tratamiento convencional con R-CHOP. Se notificaron respuestas anecdóticas a inhibidores de ALK como lorlatinib o alectinib.[Nivel de evidencia C3] Es probable que la coexpresión de CD20 y CD30 defina a un subgrupo de pacientes con LDCBG con una firma molecular única y un pronóstico más favorable. Los pacientes en este subgrupo pueden recibir un tratamiento anti-CD30 específico, como brentuximab vedotina. Los pacientes con LDCBG sin complicaciones después de 2 años tienen una SG posterior equivalente a la de la población general emparejada por edad y sexo.
Profilaxis del sistema nervioso central
El CNS-IPI es un instrumento usado para predecir qué pacientes tienen un riesgo mayor al 10 % de recidiva en el sistema nervioso central (SNC). El German Lymphoma Study Group formuló este instrumento y se validó en la base de datos de British Columbia Cancer Agency. Se usó la presencia de 4 a 6 de los factores de riesgo del CNS-IPI (edad >60 años, estado funcional ≥2, concentración elevada de LDH, enfermedad en estadio III o IV, >1 sitio extraganglionar o compromiso renal o suprarrenal) para definir el grupo de riesgo alto para la recaída en el SNC (un riesgo del 12 % de compromiso del SNC al cabo de 2 años).
Por lo general, se recomienda la profilaxis del SNC (a menudo con 4 a 6 dosis de metotrexato intratecal) para los pacientes con compromiso testicular.[Nivel de evidencia C3] En un análisis retrospectivo de los estudios alemanes RICOVER, se comparó el metotrexato intratecal con la ausencia de profilaxis en los pacientes con LDCBG. Este estudio se llevó a cabo durante la era del tratamiento con R-CHOP. Con la posible excepción de los pacientes con compromiso testicular, el análisis mostró que el metotrexato intratecal no disminuyó el riesgo de enfermedad en el SNC.[Nivel de evidencia C3] Algunos profesionales clínicos emplean dosis altas intravenosas (IV) (a menudo 4 dosis) de metotrexato en lugar del tratamiento intratecal porque esto mejora la administración del fármaco y reduce la morbilidad del paciente. En un estudio retrospectivo se evaluaron 1162 pacientes de 21 centros académicos de los Estados Unidos que recibieron metotrexato, el 77 % por vía intratecal, el 20 % dosis altas intravenosas y el 3 % por ambas vías de administración en forma secuencial (debido a los efectos tóxicos). No se observó diferencia en las tasas de recaída en el SNC entre los pacientes que recibieron metotrexato por vía intratecal y los que recibieron dosis altas intravenosas (5,4 vs. 6,8 %, P = 0,40). El compromiso testicular, el subtipo que no afecta el centro germinal y el compromiso extraganglionar alto fueron factores pronósticos del aumento de la recaída en el SNC con independencia de la vía de administración de la profilaxis. En 2 estudios retrospectivos, en los que se evaluaron las dosis altas de metotrexato en pacientes con riesgo alto de LDCBG, tampoco se indicó una mejora de la tasa de recidiva en el SNC.[Nivel de evidencia C3] Los pacientes que se consideran de riesgo alto para la recidiva en el SNC (por ejemplo, pacientes con enfermedad testicular, renal o suprarrenal, y 3 o más sitios extraganglionares) con frecuencia reciben metotrexato intratecal o dosis altas IV de metotrexato, pero la falta de estudios aleatorizados confirmatorios pone en entredicho este estándar y refleja una necesidad importante de tratamientos mejor verificados en los ensayos clínicos. Aunque no hay evidencia suficiente que respalde un beneficio significativo de la profilaxis del SNC en la mayoría de los pacientes de riesgo alto, el riesgo percibido de no tratar la recaída en el SNC a menudo ha tenido más importancia que la falta de evidencia de su eficacia. Los pacientes con compromiso testicular son una excepción, ya que en ellos se observa un beneficio con el metotrexato intratecal o las dosis altas IV de metotrexato.[Nivel de evidencia C3]
En análisis retrospectivos, la adición de rituximab a los regímenes a base de ciclofosfamida, doxorrubicina, vincristina y prednisona (CHOP) redujo de modo significativo el riesgo de recaída en el SNC.[Nivel de evidencia C3] Los pacientes con diseminación al SNC en el momento del diagnóstico o de una recaída, a menudo reciben rituximab y dosis altas de metotrexato solo o con citarabina, seguidas por TCM autógeno, pero este abordaje no se ha evaluado en ensayos aleatorizados.[Nivel de evidencia C3]
Linfoma mediastínico primario de células B grandes
El linfoma mediastínico (tímico) primario de células B grandes es un subtipo de LDCBG con características moleculares que son más similares al linfoma de Hodgkin (LH) con esclerosis nodular. Los linfomas mediastínicos con características intermedias entre linfoma mediastínico primario de células B y LH con esclerosis nodular se llaman linfomas mediastínicos de la zona gris. Por lo general, los pacientes son mujeres jóvenes (mediana de edad, 30–40 años). Los pacientes presentan una masa mediastínica anterior localmente invasiva que a veces produce síntomas respiratorios o síndrome de la vena cava superior.
El pronóstico y el tratamiento son los mismos que para otros pacientes de LDCBG en estadio comparable. En estudios no controlados de fase II en los que se emplean dosis ajustadas de R-EPOCH (etopósido, prednisona, vincristina, ciclofosfamida y doxorrubicina con rituximab) o R-CHOP, se observaron tasas de curación altas y se evitó la irradiación mediastínica.[Nivel de evidencia C1] Estos resultados indican que los pacientes que reciben regímenes a base de R-CHOP quizás eviten las complicaciones graves y a largo plazo de la radioterapia cuando se administra con quimioterapia. El uso de tomografía por emisión de positrones con flúor F 18-fludesoxiglucosa (18F-FDG) y tomografía computarizada (TEP-TC) es polémico; todavía no está claro si la TEP permite identificar de manera confiable a los pacientes que deben recibir radioterapia de consolidación o que deben omitirla.
En una revisión retrospectiva de 109 pacientes con linfoma mediastínico primario de células B grandes se indicó que el 63 % de ellos tenía una TEP-TC negativa al final del tratamiento (puntaje de Deauville 1–3). No se ofreció radioterapia, la tasa de tiempo hasta la progresión a 5 años (similar a la supervivencia sin enfermedad, pero restringida a la recaída del linfoma) fue del 90 % y la tasa de SG a 5 años fue del 97 %.[Nivel de evidencia C3] Los pacientes con una TEP-TC positiva al final del tratamiento recibieron radioterapia de consolidación. En este estudio, no está claro si esos pacientes hubieran evolucionado igual de bien sin radioterapia. Los profesionales clínicos tienen la posibilidad de vigilar la mejora de los pacientes a lo largo del tiempo con el uso de los puntajes de Deauville 4 en las imágenes de TEP-TC al final del tratamiento como alternativa a la radioterapia. Sin embargo, este abordaje no se ha estudiado en ningún ensayo clínico.
Cuando la radioterapia mediastínica abarcará el lado izquierdo del corazón o se sabe que aumentará el riesgo de cáncer de mama en pacientes jóvenes, es posible considerar el uso de terapia con protones para reducir la dosis de radiación dirigida a los órganos en riesgo. Para obtener más información, consultar la sección Síndrome de la vena cava superior en Síndromes cardiopulmonares.
Debido a que el linfoma mediastínico primario de células B grandes se caracteriza por una expresión alta del ligando 1 de muerte programada (PD-L1) y una expresión variable de CD30, en un estudio de fase II se evaluó nivolumab con brentuximab vedotina en 30 pacientes con recaída de la enfermedad. Al cabo de una mediana de seguimiento de 11,1 meses, la tasa de respuesta objetiva fue del 73 % (IC 95 %, 54−88 %).[Nivel de evidencia C3] De manera similar, en un ensayo de fase II sobre pembrolizumab en 53 pacientes con enfermedad recidivante o resistente al tratamiento, se observó una tasa de respuesta objetiva del 41,5 %. Al cabo de una mediana de seguimiento de 48,7 meses, la tasa de SSP a 4 años fue del 33,0 % y la tasa de SG a 4 años del 45,3 %.[Nivel de evidencia C3] Los 11 pacientes que lograron una respuesta completa, la seguían manteniendo en el momento del análisis final.
Entre los pacientes que habían recibido dos líneas de tratamiento previas, más de la mitad de los que recibieron terapia de células T con CAR con lisocabtagén maraleucel presentaron respuesta de la enfermedad.[Nivel de evidencia C3]
Linfoma intravascular de células B grandes (linfomatosis intravascular)
La linfomatosis intravascular se caracteriza como un linfoma de células grandes limitado al lumen intravascular. En la linfomatosis intravascular, los órganos que más a menudo están comprometidos son el encéfalo, los riñones, los pulmones y la piel.
El pronóstico es similar al del LDCBG convencional en estadio IV cuando se administra una quimioterapia combinada intensiva a base de R-CHOP, como se usa en casos de LDCBG.
Linfoma folicular (grado 3b)
Pronóstico
La evolución natural del linfoma folicular de células grandes es polémica. Si bien hay acuerdo acerca del número significativo de sobrevivientes sin enfermedad a largo plazo que tenían una enfermedad en estadio temprano, la posibilidad de curación de los pacientes con enfermedad avanzada (estadio III o estadio IV) permanece incierta. Algunos grupos notifican una tasa de recaída continua semejante a la de otros linfomas foliculares (patrón de un linfoma de crecimiento lento). Otros investigadores notifican una estabilización del tiempo sin progresión hasta los niveles previstos para un linfoma de crecimiento rápido (40 % a 10 años). Las causas de esta discrepancia quizás sean las variaciones en la clasificación histológica entre diferentes instituciones y el número escaso de pacientes con linfoma folicular de células grandes. En una revisión retrospectiva de 252 pacientes, todos tratados con quimioterapia combinada con antraciclinas, se observó que los pacientes con más del 50 % de componentes difusos en la biopsia tuvieron una SG mucho más precaria que otros pacientes de linfoma folicular de células grandes.
Abordajes terapéuticos
El tratamiento del linfoma folicular de células grandes se parece más al tratamiento del LNH de crecimiento rápido que al tratamiento del LNH de crecimiento lento. Este abordaje se sustenta en el hecho de que el tratamiento con quimioterapia de dosis alta y trasplante de células madre hematopoyéticas periféricas autógenas tiene el mismo potencial curativo en pacientes con linfoma folicular de células grandes o linfoma difuso de células grandes, que recaen.[Nivel de evidencia C1]
Entre los pacientes que habían recibido dos líneas de tratamiento previas, más de la mitad de los que recibieron terapia de células T con CAR con lisocabtagén maraleucel presentaron respuesta de la enfermedad.[Nivel de evidencia C3]
Linfoma de células de manto
El linfoma de células de manto (LCM) se encuentra en los ganglios linfáticos, el bazo, la médula ósea, la sangre, y a veces en el tubo gastrointestinal (poliposis linfomatosa). El LCM se caracteriza por células B foliculares de manto CD5+, una translocación de los cromosomas 11 y 14, así como la sobreexpresión de la proteína ciclina D1. El LCM se divide en dos subtipos clínicos. El primero es una versión clásica con linfadenopatía y expresión alta de SOX-11 que se manifiesta con una evolución clínica más maligna y un pronóstico más precario. El segundo es una versión leucémica no ganglionar con expresión baja de SOX-11, una evolución clínica menos maligna y un mejor pronóstico. Con un cariotipo complejo se predicen respuestas precarias a la terapia de inducción y una supervivencia inferior. Con frecuencia hay superposición del cuadro clínico inicial de estos subtipos, y las implicaciones terapéuticas no son evidentes. Sin embargo, ambas versiones pueden converger en la evolución y convertirse en un fenotipo blastoide o resistente al tratamiento debido a la inestabilidad y selección genómicas. Las mutaciones en TP53 se relacionan de manera especial con la progresión temprana de la enfermedad y con la muerte en los pacientes que reciben quimioinmunoterapia convencional e inhibidores de la tirosina–cinasa de Bruton (BTK) de segunda línea.
Al igual que los linfomas de grado bajo, el LCM es incurable con quimioterapia a base de antraciclinas y se presenta en pacientes de más edad con una enfermedad avanzada que por lo general es asintomática. Sin embargo, la mediana de supervivencia es significativamente más corta (5–7 años) que la de otros linfomas. Este tipo histológico ahora se considera un linfoma de crecimiento rápido (agresivo o muy maligno). El patrón difuso y la variante blastoide indican una evolución más maligna y una supervivencia más corta; mientras que el tipo de la zona de manto quizás tenga una evolución menos maligna. Es posible que una tasa de proliferación celular elevada (aumento de Ki-67, índice mitótico y microglobulina β2) se relacione con un pronóstico más precario.
Abordajes terapéuticos
Los pacientes asintomáticos con puntajes de riesgo bajo en el IPI quizás evolucionen bien cuando se difiere el tratamiento inicial.[Nivel de evidencia C3] No hay un abordaje estándar para el LCM. Se han usado varios regímenes de quimioterapia de inducción para la enfermedad progresiva sintomática. Estos regímenes varían en intensidad: rituximab solo, rituximab con ibrutinib, rituximab y bendamustina, R-CHOP o regímenes intensivos de dosis altas, como los de dosis hiperfraccionadas de CVAD (ciclofosfamida, vincristina, doxorrubicina y dexametasona alternada con metotrexato y citarabina). Después de estos regímenes, algunos médicos usan un TCM autógeno o alogénico para la consolidación, mientras que otros prefieren el mantenimiento con rituximab y reservan la consolidación con dosis altas para otro momento. El ibrutinib, la lenalidomida y el bortezomib mostraron actividad en pacientes en recaída, y ahora estos medicamentos se incorporan al tratamiento inicial.
No está claro qué abordaje terapéutico se traduce en la mejor supervivencia a largo plazo para esta entidad clinicopatológica.
En un ensayo de fase II que incluyó pacientes con LCM mayores de 64 años sin tratamiento previo, 50 pacientes recibieron el inhibidor del receptor de células B ibrutinib con rituximab. Al cabo de una mediana de seguimiento de 45 meses, la tasa de respuesta general fue del 96 %, la tasa de respuesta completa fue del 71 %, la tasa de SSP a 3 años fue del 87 % y la tasa de SG a 3 años fue del 94 %.[Nivel de evidencia C3] En un ensayo de fase II de 131 pacientes de 65 años o menos con LCM sin tratamiento previo, la administración durante 1 año de ibrutinib y 4 semanas de rituximab, produjo una tasa de respuesta completa del 89 % antes de cualquier consolidación con quimioterapia.[Nivel de evidencia C3] En otro ensayo de fase II con ibrutinib y rituximab se incluyó a pacientes asintomáticos con LCM sin tratamiento previo; la tasa de respuesta completa fue del 87 %.[Nivel de evidencia C3] Los pacientes con tratamiento previo que recibieron ibrutinib tuvieron una tasa de respuesta del 86 % (tasa de respuesta completa del 21 %) y una mediana de SSP de 14 meses.[Nivel de evidencia C3] En un ensayo prospectivo aleatorizado, 280 pacientes de LCM en recaída o resistente al tratamiento recibieron ibrutinib o temsirólimus. Después de una mediana de seguimiento de 15 meses, la mediana de SSP favoreció al ibrutinib (14,6 vs. 6,2 meses; cociente de riesgos instantáneos [CRI], 0,43; IC 95 %, 0,32–0,58, P < 0,001).[Nivel de evidencia B1] En un estudio de fase II con 23 pacientes de LCM en recaída o resistente al tratamiento, se combinó el ibrutinib con otro fármaco activo, venetoclax. Se obtuvieron resultados sin precedentes: un 71 % de los pacientes tuvo una respuesta completa y, de ellos, el 78 % mantuvo la respuesta al cabo de 15 meses.[Nivel de evidencia C3]
En un ensayo aleatorizado prospectivo se incluyeron 523 pacientes de 65 años o más con LCM. Los pacientes se asignaron de manera aleatoria para recibir ibrutinib, bendamustina y rituximab o bendamustina y rituximab solos. Después de una mediana de seguimiento de 84,7 meses, la mediana de SSP fue de 80,6 meses para los pacientes que recibieron ibrutinib y de 52,9 meses para los pacientes que recibieron bendamustina y rituximab solos (CRI, 0,75; IC 95 %, 0,59–0,96; P = 0,01). No hubo diferencia en la tasa de SG a 7 años (55,0 vs. 56,8 %; CRI, 1,07; IC 95 %, 0,81–1,40).[Nivel de evidencia B1] No está claro si los pacientes que reciben ibrutinib solo, podrían lograr estos mismos resultados sin recibir quimioterapia convencional. La magnitud del beneficio que se observó en los resultados para la SSP contrastó con el beneficio insuficiente para la SG después de 7 años lo que podría poner en duda la inocuidad a largo plazo de esta combinación.
En un ensayo prospectivo aleatorizado, 560 pacientes mayores de 60 años que no eran aptos para un TCM recibieron R-CHOP o R-FC (rituximab, fludarabina y ciclofosfamida) durante 6 a 8 ciclos, luego los pacientes que respondieron se asignaron en forma aleatoria a terapia de mantenimiento con rituximab o interferón α. Con una mediana de seguimiento de 7,6 años, la mediana de SG fue significativamente más corta después de R-FC que después de R-CHOP (3,9 vs. 6,4 años; P = 0,0054).[Nivel de evidencia A1] En el mismo ensayo, con una mediana de seguimiento de 8 años para los 316 pacientes que respondieron, el mantenimiento con rituximab mejoró la SG en comparación con el mantenimiento con interferón (mediana de SG, 9,8 vs. 7,1 años; P = 0,009).[Nivel de evidencia A1] Los pacientes que respondieron a R-CHOP se beneficiaron más del rituximab en la SG (mediana, 9,8 vs. 6,4 años; P = 0,0026).[Nivel de evidencia C1] En un ensayo aleatorizado, se comparó bendamustina y rituximab (BR) con R-CHOP y se observó una mejora de la SSP (35 vs. 22 meses; CRI, 0,49; IC 95 %, 0,28–0,79; P = 0,004), pero ninguna diferencia en la SG.[Nivel de evidencia B1] Sin embargo, en este ensayo no se observó ningún beneficio para el mantenimiento del rituximab después de BR. En un ensayo prospectivo aleatorizado con 487 pacientes, se comparó un régimen de bortezomib, rituximab, ciclofosfamida, doxorrubicina y prednisona (VR-CAP) con R-CHOP. Al cabo de una mediana de seguimiento de 82 meses, la mediana de SG fue superior para VR-CAP (90,7 meses) en comparación con R-CHOP (55,7 meses) (CRI, 0,66; IC 95 %, 0,51−0,85; P = 0,001).[Nivel de evidencia A1]
En un ensayo prospectivo aleatorizado con 497 pacientes menores de 65 años se compararon 6 ciclos de R-CHOP y 6 ciclos alternados de R-CHOP con rituximab, dexametasona, citarabina y cisplatino (R-DHAP); ambos grupos se sometieron a TCM autógeno.[Nivel de evidencia B1] Al cabo de una mediana de seguimiento de 10,6 años, la tasa de SSP a 10 años fue del 73 % en los pacientes que recibieron R-DHAP y del 57 % en los que recibieron R-CHOP (CRI, 0,56; P = 0,038), pero no hubo diferencias en las tasas de SG a 10 años (60 % [R-DHAP] vs. 55 % [R-CHOP]; CRI, 0,80; 0,61–1,06; P = 0,12).[Nivel de evidencia B1] Este es el ensayo aleatorizado al que se hace referencia en todos los artículos subsiguientes en el que se establece una función de la citarabina en la terapia de inducción; la falta definitiva de ventaja en la supervivencia pone en duda esta afirmación.
En los ensayos aleatorizados no se han confirmado los beneficios para la SG en los pacientes que reciben terapia de consolidación con TCM autógenodesde la introducción del rituximab. En un análisis retrospectivo de 1265 pacientes menores de 65 años aptos para trasplante no se observó ningún beneficio del TCM autógeno durante el tiempo que transcurrió hasta el siguiente tratamiento (CRI, 0,84; IC 95 %, 0,68–1,03) o SG (CRI, 0,86; IC 95 %, 0,63–1,18).[Nivel de evidencia C3] En este mismo análisis retrospectivo de cohortes del mundo real se halló un beneficio para el rituximab de mantenimiento (tras la inducción de BR) durante el tiempo transcurrido hasta el siguiente tratamiento (CRI, 1,96; IC 95 %, 1,61–2,38; P< 0,001) y la SG (CRI, 1,51; IC 95 %, 1,19–1,92; P < 0,001).[Nivel de evidencia C3]
En un ensayo prospectivo (NCT00921414) de 299 pacientes con LCM sin tratamiento previo, 257 pacientes que respondieron recibieron 4 ciclos de R-DHAP y TCM autógeno. Los pacientes se asignaron de manera aleatoria para recibir terapia de mantenimiento con rituximab por 3 años versus ninguna terapia de mantenimiento. Después de la aleatorización y tras una mediana de seguimiento de 50,2 meses, se observaron tasas de SSP a 4 años en el grupo de mantenimiento con rituximab del 83 % (IC 95 %, 73–88 %) y en el grupo que no recibió mantenimiento del 64 % (IC 95 %, 55–73 %; P < 0,001). La tasa de SG a 4 años del 89 % (IC 95 %, 81–94 %) también favoreció al grupo de mantenimiento con rituximab versus la del 80 % (IC 95 %, 72–88 %; P = 0,04) en el grupo que no recibió mantenimiento.[Nivel de evidencia A1]
La lenalidomida con rituximab o sin este también produce tasas de respuesta de alrededor del 50 % en pacientes en recaída, y tasas de respuesta incluso más altas en pacientes sin tratamiento previo.[Nivel de evidencia C3]
El acalabrutinib (otro inhibidor del receptor de células B de la vía de la BTK) se estudió en 124 pacientes con LCM en recaída o resistente al tratamiento. En un estudio de fase II, la tasa de respuesta general fue del 81 %, la tasa de respuesta completa fue del 40 %, y la tasa de SSP a 1 año fue del 67 %.[Nivel de evidencia C3] Asimismo, en un estudio de fase II de 86 pacientes con LCM en recaída o resistente al tratamiento, se evaluó el inhibidor de la BTK, zanubrutinib. Después de una mediana de seguimiento de 35,3 meses, la tasa de respuesta general fue del 84 %, la tasa de respuesta completa fue del 78 % y la mediana de SSP fue de 33,0 meses.[Nivel de evidencia C3]
Los pacientes con LCM en recaída o resistente al tratamiento cuya enfermedad no respondió al ibrutinib o acalabrutinib se inscribieron en un ensayo de fase II (ZUMA-2 [NCT02601313]) de brexucabtagén autoleucel, una terapia de células T con CAR anti-CD19. Al cabo de una mediana de seguimiento de 36 meses, 68 pacientes tuvieron una tasa de respuesta objetiva del 91 % (IC 95 %, 82−97 %) y una tasa de respuesta completa del 68 % (IC 95 %, 55−78 %). La mediana de SSP y SG fue de 25,8 meses (IC 95 %, 10–48) y de 46,6 meses (IC 95 %, 24,9–no estimable), respectivamente.[Nivel de evidencia C3] De los pacientes, el 15 % presentaron un síndrome de liberación de citocinas de grado 3 o superior, y el 31 % de los pacientes presentaron complicaciones neurológicas. En una evaluación retrospectiva posterior de 16 instituciones se incluyeron 168 pacientes que recibieron brexucabtagén autoleucel como parte del U.S. Lymphoma CAR-T Consortium. En el estudio se observaron resultados similares a los del ensayo ZUMA-2.[Nivel de evidencia C3]
El pirtobrutinib, inhibidor reversible y no covalente de la BTK, se evaluó en un estudio de fase I/II de 164 pacientes con LCM. De los pacientes, 79 (87,8 %) recibieron al menos una dosis recomendada de la fase II de 200 mg una vez al día. Entre los 90 pacientes tratados previamente con inhibidores covalentes de la BTK incluidos en la cohorte de eficacia primaria, la tasa de respuesta general fue del 57,8 % (IC 95 %, 46,9–68,1%), incluida una tasa de respuesta completa del 20,0 %. Al cabo de una mediana de seguimiento de 12 meses, la duración mediana de la respuesta fue de 21,6 meses (IC 95 %, 7,5–no alcanzada). En la cohorte de seguridad de 164 pacientes con LCM, los eventos adversos más frecuentes que aparecieron durante el tratamiento fueron fatiga (29,9 %), diarrea (21,3 %) y disnea (16,5 %). Se produjeron hemorragias de grado 3 o superior en el 3,7 % de los pacientes y fibrilación o aleteo auricular en el 1,2 %. Solo el 3 % de los pacientes interrumpió el tratamiento con pirtobrutinib debido a un evento adverso relacionado con el tratamiento.[Nivel de evidencia C3] La Administración de Alimentos y Medicamentos de los Estados Unidos aprobó el uso de pirtobrutinib en pacientes que hayan recibido al menos 2 líneas de tratamiento previas, incluso otro inhibidor de la BTK.
En resumen, la secuenciación óptima de estas terapias diversas no está clara y es objeto de un ensayo clínico intergrupal en curso. El rituximab, la lenalidomida, el ibrutinib, el acalabrutinib, el zanubrutinib, el pirtobrutinib y el venetoclax son fármacos biológicos directos que quizás conduzcan a estrategias de tratamiento sin quimioterapia para los pacientes con LCM. La mayoría de los estudios apoyan el uso de un tratamiento de mantenimiento con rituximab durante 2 a 3 años tras la terapia de inducción (con o sin consolidación).
Nunca se ha estudiado en un ensayo prospectivo aleatorizado la administración rutinaria de profilaxis del SNC para el LCM de riesgo alto. El uso de metotrexato intratecal o de dosis altas y el uso de terapias sistémicas con penetración en el SNC, como el ibrutinib, la citarabina en dosis altas o el venetoclax, no se ha estudiado ni se ha comprobado su eficacia en esta situación.
Linfoma de Burkitt o linfoma difuso de células pequeñas no hendidas
El linfoma de Burkitt o el linfoma difuso de células pequeñas no hendidas por lo general afecta a pacientes más jóvenes y es el tipo más común de LNH infantil. Estos tipos de linfomas de células B de crecimiento rápido (agresivos) extraganglionares se caracterizan por la translocación y desregulación del gen MYC en el cromosoma 8. Un subgrupo de pacientes con translocación doble de MYC y BCL2 tuvieron un desenlace muy precario a pesar del tratamiento intensivo (mediana de SG, 5 meses).[Nivel de evidencia C1]
En algunos pacientes con células B más grandes hay una superposición morfológica con el LDCBG. Estos linfomas de células grandes de tipo Burkitt exhiben una desregulación de MYC, tasas de proliferación muy altas y un perfil de expresión génica compatible con el linfoma de Burkitt clásico. En casos endémicos, a menudo en África, hay compromiso de los huesos faciales o los maxilares en los niños, y en su mayoría contienen genomas del virus de Epstein-Barr (VEB). Los casos esporádicos suelen afectar el aparato digestivo, los ovarios o los riñones. Los pacientes que presentan masas de crecimiento rápido y una concentración muy alta de LDH, se podrían curar con quimioterapia combinada intensiva a base de doxorrubicina.
Abordajes terapéuticos
El tratamiento del linfoma de Burkitt o el linfoma difuso de células pequeñas no hendidas incluye regímenes multifarmacológicos intensivos en combinación con rituximab, similares a los que se usan para los linfomas de crecimiento rápido en estadios avanzados (linfomas difusos de células grades). Una quimioterapia combinada intensiva, como la que se utiliza para el linfoma de Burkitt infantil resultó muy exitosa para los pacientes adultos con enfermedad en estadio avanzado que lograron una supervivencia sin enfermedad a 5 años de más del 60 %. Los factores de pronóstico adverso son la enfermedad con gran masa tumoral abdominal y una concentración sérica de LDH alta. Los pacientes con linfoma de Burkitt tienen un riesgo de por vida del 20 % al 30 % de compromiso del SNC. Se necesita profilaxis con quimioterapia intratecal durante la terapia de inducción. Los pacientes con linfoma de Burkitt relacionado con el virus de la inmunodeficiencia humana (VIH) también se benefician de una modificación menos tóxica de los regímenes multifarmacológicos intensivos en combinación con rituximab.[Nivel de evidencia C3] Para obtener más información, consultar Tratamiento del linfoma primario del sistema nervioso central y Tratamiento del linfoma relacionado con el SIDA.
Linfoma linfoblástico de células B
El linfoma linfoblástico de células B (células T precursoras) es una forma muy maligna de LNH, que también se conoce como linfoma linfoblástico B o LLB-B. Es menos común que el linfoma linfoblástico de células T.
El tratamiento por lo general es similar al de la leucemia linfoblástica aguda. La quimioterapia combinada intensiva con trasplante de médula ósea o sin este es el tratamiento estándar para este tipo histológico muy maligno o agresivo de LNH. A veces, se administra radioterapia dirigida a los lugares con gran masa tumoral (tumor voluminoso). Debido a que estas formas de LNH tienden a progresar rápido, la quimioterapia combinada se administra de inmediato una vez que se confirma el diagnóstico. Los aspectos más importantes de la evaluación de estadificación antes del tratamiento incluyen una revisión minuciosa de las piezas patológicas, las muestras de aspirado de la médula ósea, las muestras de la biopsia, las pruebas citológicas del líquido cefalorraquídeo y de los marcadores de linfocitos. Para obtener más información, consultar Tratamiento de la leucemia linfoblástica aguda en adultos.
Linfoma primario de efusiones
El linfoma primario de efusiones casi siempre ocurre en la cavidad pleural, pericárdica o abdominal en ausencia de una masa tumoral identificable. A menudo los pacientes son seropositivos para el VIH y, por lo general, el tumor contiene el virus del herpes relacionado con el sarcoma de Kaposi o el virus del herpes humano de tipo 8.
Pronóstico
El pronóstico para el linfoma primario de efusiones es muy precario.
Abordajes terapéuticos
El tratamiento suele ser similar al de los linfomas difusos de células grandes en estadio comparable.
Linfoma plasmoblástico
El linfoma plasmoblástico (plasmablástico) se observa con más frecuencia en pacientes con infección por el VIH y suele contener células B grandes CD20 con características plasmacíticas. Este tipo de linfoma tiene una evolución clínica muy maligna, con respuestas precarias y remisiones cortas a la quimioterapia estándar. Hay informes anecdóticos en los que se indica que a veces es posible administrar la quimioterapia intensiva que se usa para el linfoma de Burkitt o el linfoma linfoblástico, seguida de consolidación con TCM en los pacientes que responden a esta.
Trastorno linfoproliferativo postrasplante polimórfico
Los pacientes sometidos a trasplante de corazón, pulmón, hígado, riñón o páncreas suelen necesitar inmunodepresión de por vida. La inmunodepresión a veces produce un trastorno linfoproliferativo postrasplante (TLPT) en el 1 % al 3 % de los receptores; el cual es similar a un linfoma de crecimiento rápido. Los patólogos pueden diferenciar la hiperplasia policlonal de células B y el linfoma monoclonal de células B que casi siempre se relacionan con el VEB.
Pronóstico
Los factores de pronóstico precario del TLPT son el estado funcional deficiente, el compromiso del órgano injertado, un IPI alto, una LDH elevada y enfermedad en múltiples sitios.
Opciones de tratamiento
En algunos casos la interrupción del tratamiento inmunodepresor lleva a la erradicación del linfoma. Cuando esto no es exitoso ni posible, se considera el uso de rituximab, ya que produjo remisiones duraderas en alrededor de un 60 % de los pacientes y tiene un perfil de toxicidad favorable. Si estas medidas fracasan, se recomienda la quimioterapia combinada a base de doxorrubicina (R-CHOP), aunque en algunos pacientes es posible evitar el tratamiento citotóxico. Las presentaciones localizadas se pueden controlar con cirugía o radioterapia sola. Estas masas localizadas, que a veces crecen durante meses, a menudo tienen un fenotipo policlonal y tienden a surgir en el transcurso de semanas o pocos meses tras el trasplante. La enfermedad multifocal rápidamente progresiva se presenta de manera más tardía después del trasplante (>1 año), a menudo tiene un fenotipo monoclonal y se relaciona con el VEB. Estos pacientes en ocasiones presentan remisiones prolongadas con los regímenes de quimioterapia estándar que se usan para el linfoma de crecimiento rápido. Los casos de TLPT negativo para VEB se presentan todavía más tarde (mediana de 5 años después del trasplante) y tienen un pronóstico más precario; en estas circunstancias la administración directa de quimioterapia R-CHOP es posible. Se obtuvo una respuesta clínica sostenida con una inmunotoxina (un antígeno de superficie de las células B anti-CD22 unido a ricina, una toxina vegetal) después del fracaso de la quimioterapia. También está en evaluación clínica un anticuerpo monoclonal anti-interleucina 6.
Granulomatosis linfomatoide
La granulomatosis linfomatoide es un linfoma de células B grandes positivo para el VEB con un fondo predominante de células T. En el examen histológico se observa angioinvasión y vasculitis que a menudo se manifiestan como lesiones pulmonares o compromiso de los senos paranasales.
Los pacientes se tratan como los de linfoma difuso de células grandes y necesitan quimioterapia combinada a base de doxorrubicina.
References
- Sehn LH, Salles G: Diffuse Large B-Cell Lymphoma. N Engl J Med 384 (9): 842-858, 2021.
- Delabie J, Vandenberghe E, Kennes C, et al.: Histiocyte-rich B-cell lymphoma. A distinct clinicopathologic entity possibly related to lymphocyte predominant Hodgkin's disease, paragranuloma subtype. Am J Surg Pathol 16 (1): 37-48, 1992.
- Achten R, Verhoef G, Vanuytsel L, et al.: T-cell/histiocyte-rich large B-cell lymphoma: a distinct clinicopathologic entity. J Clin Oncol 20 (5): 1269-77, 2002.
- Bouabdallah R, Mounier N, Guettier C, et al.: T-cell/histiocyte-rich large B-cell lymphomas and classical diffuse large B-cell lymphomas have similar outcome after chemotherapy: a matched-control analysis. J Clin Oncol 21 (7): 1271-7, 2003.
- Ghesquières H, Berger F, Felman P, et al.: Clinicopathologic characteristics and outcome of diffuse large B-cell lymphomas presenting with an associated low-grade component at diagnosis. J Clin Oncol 24 (33): 5234-41, 2006.
- Miller TP, Dahlberg S, Cassady JR, et al.: Chemotherapy alone compared with chemotherapy plus radiotherapy for localized intermediate- and high-grade non-Hodgkin's lymphoma. N Engl J Med 339 (1): 21-6, 1998.
- Coiffier B, Lepage E, Briere J, et al.: CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 346 (4): 235-42, 2002.
- Coiffier B: State-of-the-art therapeutics: diffuse large B-cell lymphoma. J Clin Oncol 23 (26): 6387-93, 2005.
- Habermann TM, Weller EA, Morrison VA, et al.: Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. J Clin Oncol 24 (19): 3121-7, 2006.
- Zhou Z, Sehn LH, Rademaker AW, et al.: An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 123 (6): 837-42, 2014.
- Møller MB, Christensen BE, Pedersen NT: Prognosis of localized diffuse large B-cell lymphoma in younger patients. Cancer 98 (3): 516-21, 2003.
- Maurer MJ, Ghesquières H, Link BK, et al.: Diagnosis-to-Treatment Interval Is an Important Clinical Factor in Newly Diagnosed Diffuse Large B-Cell Lymphoma and Has Implication for Bias in Clinical Trials. J Clin Oncol 36 (16): 1603-1610, 2018.
- Scott DW, King RL, Staiger AM, et al.: High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with diffuse large B-cell lymphoma morphology. Blood 131 (18): 2060-2064, 2018.
- Horn H, Ziepert M, Becher C, et al.: MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 121 (12): 2253-63, 2013.
- Staiger AM, Ziepert M, Horn H, et al.: Clinical Impact of the Cell-of-Origin Classification and the MYC/ BCL2 Dual Expresser Status in Diffuse Large B-Cell Lymphoma Treated Within Prospective Clinical Trials of the German High-Grade Non-Hodgkin's Lymphoma Study Group. J Clin Oncol 35 (22): 2515-2526, 2017.
- Howlett C, Snedecor SJ, Landsburg DJ, et al.: Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol 170 (4): 504-14, 2015.
- Sesques P, Johnson NA: Approach to the diagnosis and treatment of high-grade B-cell lymphomas with MYC and BCL2 and/or BCL6 rearrangements. Blood 129 (3): 280-288, 2017.
- Landsburg DJ, Falkiewicz MK, Maly J, et al.: Outcomes of Patients With Double-Hit Lymphoma Who Achieve First Complete Remission. J Clin Oncol 35 (20): 2260-2267, 2017.
- Herrera AF, Mei M, Low L, et al.: Relapsed or Refractory Double-Expressor and Double-Hit Lymphomas Have Inferior Progression-Free Survival After Autologous Stem-Cell Transplantation. J Clin Oncol 35 (1): 24-31, 2017.
- Canellos GP: CHOP may have been part of the beginning but certainly not the end: issues in risk-related therapy of large-cell lymphoma. J Clin Oncol 15 (5): 1713-6, 1997.
- Sha C, Barrans S, Cucco F, et al.: Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. J Clin Oncol 37 (3): 202-212, 2019.
- Soumerai JD, Rosenthal A, Harkins S, et al.: Next-generation ALK inhibitors are highly active in ALK-positive large B-cell lymphoma. Blood 140 (16): 1822-1826, 2022.
- Hu S, Xu-Monette ZY, Balasubramanyam A, et al.: CD30 expression defines a novel subgroup of diffuse large B-cell lymphoma with favorable prognosis and distinct gene expression signature: a report from the International DLBCL Rituximab-CHOP Consortium Program Study. Blood 121 (14): 2715-24, 2013.
- Maurer MJ, Ghesquières H, Jais JP, et al.: Event-free survival at 24 months is a robust end point for disease-related outcome in diffuse large B-cell lymphoma treated with immunochemotherapy. J Clin Oncol 32 (10): 1066-73, 2014.
- Schmitz N, Zeynalova S, Nickelsen M, et al.: CNS International Prognostic Index: A Risk Model for CNS Relapse in Patients With Diffuse Large B-Cell Lymphoma Treated With R-CHOP. J Clin Oncol 34 (26): 3150-6, 2016.
- Zucca E, Conconi A, Mughal TI, et al.: Patterns of outcome and prognostic factors in primary large-cell lymphoma of the testis in a survey by the International Extranodal Lymphoma Study Group. J Clin Oncol 21 (1): 20-7, 2003.
- Vitolo U, Chiappella A, Ferreri AJ, et al.: First-line treatment for primary testicular diffuse large B-cell lymphoma with rituximab-CHOP, CNS prophylaxis, and contralateral testis irradiation: final results of an international phase II trial. J Clin Oncol 29 (20): 2766-72, 2011.
- Cheah CY, Wirth A, Seymour JF: Primary testicular lymphoma. Blood 123 (4): 486-93, 2014.
- Boehme V, Schmitz N, Zeynalova S, et al.: CNS events in elderly patients with aggressive lymphoma treated with modern chemotherapy (CHOP-14) with or without rituximab: an analysis of patients treated in the RICOVER-60 trial of the German High-Grade Non-Hodgkin Lymphoma Study Group (DSHNHL). Blood 113 (17): 3896-902, 2009.
- Glantz MJ, Cole BF, Recht L, et al.: High-dose intravenous methotrexate for patients with nonleukemic leptomeningeal cancer: is intrathecal chemotherapy necessary? J Clin Oncol 16 (4): 1561-7, 1998.
- Orellana-Noia VM, Reed DR, McCook AA, et al.: Single-route CNS prophylaxis for aggressive non-Hodgkin lymphomas: real-world outcomes from 21 US academic institutions. Blood 139 (3): 413-423, 2022.
- Puckrin R, El Darsa H, Ghosh S, et al.: Ineffectiveness of high-dose methotrexate for prevention of CNS relapse in diffuse large B-cell lymphoma. Am J Hematol 96 (7): 764-771, 2021.
- Jeong H, Cho H, Kim H, et al.: Efficacy and safety of prophylactic high-dose MTX in high-risk DLBCL: a treatment intent-based analysis. Blood Adv 5 (8): 2142-2152, 2021.
- Eyre TA, Savage KJ, Cheah CY, et al.: CNS prophylaxis for diffuse large B-cell lymphoma. Lancet Oncol 23 (9): e416-e426, 2022.
- Villa D, Connors JM, Shenkier TN, et al.: Incidence and risk factors for central nervous system relapse in patients with diffuse large B-cell lymphoma: the impact of the addition of rituximab to CHOP chemotherapy. Ann Oncol 21 (5): 1046-52, 2010.
- Ferreri AJ, Donadoni G, Cabras MG, et al.: High Doses of Antimetabolites Followed by High-Dose Sequential Chemoimmunotherapy and Autologous Stem-Cell Transplantation in Patients With Systemic B-Cell Lymphoma and Secondary CNS Involvement: Final Results of a Multicenter Phase II Trial. J Clin Oncol 33 (33): 3903-10, 2015.
- Schmitz N, Wu HS: Advances in the Treatment of Secondary CNS Lymphoma. J Clin Oncol 33 (33): 3851-3, 2015.
- Savage KJ: Primary mediastinal large B-cell lymphoma. Blood 140 (9): 955-970, 2022.
- van Besien K, Kelta M, Bahaguna P: Primary mediastinal B-cell lymphoma: a review of pathology and management. J Clin Oncol 19 (6): 1855-64, 2001.
- Dunleavy K, Wilson WH: Primary mediastinal B-cell lymphoma and mediastinal gray zone lymphoma: do they require a unique therapeutic approach? Blood 125 (1): 33-9, 2015.
- Dunleavy K, Pittaluga S, Maeda LS, et al.: Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med 368 (15): 1408-16, 2013.
- Savage KJ, Yenson PR, Shenkier T, et al.: The outcome of primary mediastinal large B-cell lymphoma in the R-CHOP treatment era. [Abstract] Blood 120 (21): A-303, 2012.
- Vassilakopoulos TP, Pangalis GA, Katsigiannis A, et al.: Rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone with or without radiotherapy in primary mediastinal large B-cell lymphoma: the emerging standard of care. Oncologist 17 (2): 239-49, 2012.
- Rieger M, Osterborg A, Pettengell R, et al.: Primary mediastinal B-cell lymphoma treated with CHOP-like chemotherapy with or without rituximab: results of the Mabthera International Trial Group study. Ann Oncol 22 (3): 664-70, 2011.
- Gleeson M, Hawkes EA, Cunningham D, et al.: Rituximab, cyclophosphamide, doxorubicin, vincristine and prednisolone (R-CHOP) in the management of primary mediastinal B-cell lymphoma: a subgroup analysis of the UK NCRI R-CHOP 14 versus 21 trial. Br J Haematol 175 (4): 668-672, 2016.
- Held G, Thurner L, Poeschel V, et al.: Role of radiotherapy and dose-densification of R-CHOP in primary mediastinal B-cell lymphoma: A subgroup analysis of the unfolder trial of the German Lymphoma Alliance (GLA). [Abstract] J Clin Oncol 38 (Suppl 15): A-8041, 2021.
- Martelli M, Ceriani L, Zucca E, et al.: [18F]fluorodeoxyglucose positron emission tomography predicts survival after chemoimmunotherapy for primary mediastinal large B-cell lymphoma: results of the International Extranodal Lymphoma Study Group IELSG-26 Study. J Clin Oncol 32 (17): 1769-75, 2014.
- Zinzani PL, Broccoli A, Casadei B, et al.: The role of rituximab and positron emission tomography in the treatment of primary mediastinal large B-cell lymphoma: experience on 74 patients. Hematol Oncol 33 (4): 145-50, 2015.
- Ceriani L, Martelli M, Conconi A, et al.: Prognostic models for primary mediastinal (thymic) B-cell lymphoma derived from 18-FDG PET/CT quantitative parameters in the International Extranodal Lymphoma Study Group (IELSG) 26 study. Br J Haematol 178 (4): 588-591, 2017.
- Hayden AR, Tonseth P, Lee DG, et al.: Outcome of primary mediastinal large B-cell lymphoma using R-CHOP: impact of a PET-adapted approach. Blood 136 (24): 2803-2811, 2020.
- Dabaja BS, Hoppe BS, Plastaras JP, et al.: Proton therapy for adults with mediastinal lymphomas: the International Lymphoma Radiation Oncology Group guidelines. Blood 132 (16): 1635-1646, 2018.
- Zinzani PL, Santoro A, Gritti G, et al.: Nivolumab Combined With Brentuximab Vedotin for Relapsed/Refractory Primary Mediastinal Large B-Cell Lymphoma: Efficacy and Safety From the Phase II CheckMate 436 Study. J Clin Oncol 37 (33): 3081-3089, 2019.
- Zinzani PL, Thieblemont C, Melnichenko V, et al.: Pembrolizumab in relapsed or refractory primary mediastinal large B-cell lymphoma: final analysis of KEYNOTE-170. Blood 142 (2): 141-145, 2023.
- Abramson JS, Palomba ML, Gordon LI, et al.: Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 396 (10254): 839-852, 2020.
- Shimada K, Matsue K, Yamamoto K, et al.: Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol 26 (19): 3189-95, 2008.
- Ponzoni M, Ferreri AJ, Campo E, et al.: Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol 25 (21): 3168-73, 2007.
- Shimada K, Yamaguchi M, Atsuta Y, et al.: Rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone combined with high-dose methotrexate plus intrathecal chemotherapy for newly diagnosed intravascular large B-cell lymphoma (PRIMEUR-IVL): a multicentre, single-arm, phase 2 trial. Lancet Oncol 21 (4): 593-602, 2020.
- Longo DL: What's the deal with follicular lymphomas? J Clin Oncol 11 (2): 202-8, 1993.
- Anderson JR, Vose JM, Bierman PJ, et al.: Clinical features and prognosis of follicular large-cell lymphoma: a report from the Nebraska Lymphoma Study Group. J Clin Oncol 11 (2): 218-24, 1993.
- Bartlett NL, Rizeq M, Dorfman RF, et al.: Follicular large-cell lymphoma: intermediate or low grade? J Clin Oncol 12 (7): 1349-57, 1994.
- Wendum D, Sebban C, Gaulard P, et al.: Follicular large-cell lymphoma treated with intensive chemotherapy: an analysis of 89 cases included in the LNH87 trial and comparison with the outcome of diffuse large B-cell lymphoma. Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 15 (4): 1654-63, 1997.
- Hans CP, Weisenburger DD, Vose JM, et al.: A significant diffuse component predicts for inferior survival in grade 3 follicular lymphoma, but cytologic subtypes do not predict survival. Blood 101 (6): 2363-7, 2003.
- Vose JM, Bierman PJ, Lynch JC, et al.: Effect of follicularity on autologous transplantation for large-cell non-Hodgkin's lymphoma. J Clin Oncol 16 (3): 844-9, 1998.
- Cohen JB, Zain JM, Kahl BS: Current Approaches to Mantle Cell Lymphoma: Diagnosis, Prognosis, and Therapies. Am Soc Clin Oncol Educ Book 37: 512-525, 2017.
- Clot G, Jares P, Giné E, et al.: A gene signature that distinguishes conventional and leukemic nonnodal mantle cell lymphoma helps predict outcome. Blood 132 (4): 413-422, 2018.
- Greenwell IB, Staton AD, Lee MJ, et al.: Complex karyotype in patients with mantle cell lymphoma predicts inferior survival and poor response to intensive induction therapy. Cancer 124 (11): 2306-2315, 2018.
- Dreyling M, Klapper W, Rule S: Blastoid and pleomorphic mantle cell lymphoma: still a diagnostic and therapeutic challenge! Blood 132 (26): 2722-2729, 2018.
- Jain P, Dreyling M, Seymour JF, et al.: High-Risk Mantle Cell Lymphoma: Definition, Current Challenges, and Management. J Clin Oncol 38 (36): 4302-4316, 2020.
- Lew TE, Minson A, Dickinson M, et al.: Treatment approaches for patients with TP53-mutated mantle cell lymphoma. Lancet Haematol 10 (2): e142-e154, 2023.
- Herrmann A, Hoster E, Zwingers T, et al.: Improvement of overall survival in advanced stage mantle cell lymphoma. J Clin Oncol 27 (4): 511-8, 2009.
- Majlis A, Pugh WC, Rodriguez MA, et al.: Mantle cell lymphoma: correlation of clinical outcome and biologic features with three histologic variants. J Clin Oncol 15 (4): 1664-71, 1997.
- Tiemann M, Schrader C, Klapper W, et al.: Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): a clinicopathological study from the European MCL Network. Br J Haematol 131 (1): 29-38, 2005.
- Campo E, Raffeld M, Jaffe ES: Mantle-cell lymphoma. Semin Hematol 36 (2): 115-27, 1999.
- Martin P, Chadburn A, Christos P, et al.: Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 27 (8): 1209-13, 2009.
- Cohen JB, Han X, Jemal A, et al.: Deferred therapy is associated with improved overall survival in patients with newly diagnosed mantle cell lymphoma. Cancer 122 (15): 2356-63, 2016.
- Gerson JN, Handorf E, Villa D, et al.: Survival Outcomes of Younger Patients With Mantle Cell Lymphoma Treated in the Rituximab Era. J Clin Oncol 37 (6): 471-480, 2019.
- Goy A, Kalayoglu Besisik S, Drach J, et al.: Longer-term follow-up and outcome by tumour cell proliferation rate (Ki-67) in patients with relapsed/refractory mantle cell lymphoma treated with lenalidomide on MCL-001(EMERGE) pivotal trial. Br J Haematol 170 (4): 496-503, 2015.
- Ruan J, Martin P, Shah B, et al.: Lenalidomide plus Rituximab as Initial Treatment for Mantle-Cell Lymphoma. N Engl J Med 373 (19): 1835-44, 2015.
- Wang ML, Rule S, Martin P, et al.: Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 369 (6): 507-16, 2013.
- Wang ML, Blum KA, Martin P, et al.: Long-term follow-up of MCL patients treated with single-agent ibrutinib: updated safety and efficacy results. Blood 126 (6): 739-45, 2015.
- Ruan J, Martin P, Christos P, et al.: Five-year follow-up of lenalidomide plus rituximab as initial treatment of mantle cell lymphoma. Blood 132 (19): 2016-2025, 2018.
- Jain P, Zhao S, Lee HJ, et al.: Ibrutinib With Rituximab in First-Line Treatment of Older Patients With Mantle Cell Lymphoma. J Clin Oncol 40 (2): 202-212, 2022.
- Wang ML, Jain P, Zhao S, et al.: Ibrutinib-rituximab followed by R-HCVAD as frontline treatment for young patients (≤65 years) with mantle cell lymphoma (WINDOW-1): a single-arm, phase 2 trial. Lancet Oncol 23 (3): 406-415, 2022.
- Giné E, de la Cruz F, Jiménez Ubieto A, et al.: Ibrutinib in Combination With Rituximab for Indolent Clinical Forms of Mantle Cell Lymphoma (IMCL-2015): A Multicenter, Open-Label, Single-Arm, Phase II Trial. J Clin Oncol 40 (11): 1196-1205, 2022.
- Dreyling M, Jurczak W, Jerkeman M, et al.: Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet 387 (10020): 770-8, 2016.
- Tam CS, Anderson MA, Pott C, et al.: Ibrutinib plus Venetoclax for the Treatment of Mantle-Cell Lymphoma. N Engl J Med 378 (13): 1211-1223, 2018.
- Wang ML, Jurczak W, Jerkeman M, et al.: Ibrutinib plus Bendamustine and Rituximab in Untreated Mantle-Cell Lymphoma. N Engl J Med 386 (26): 2482-2494, 2022.
- Kluin-Nelemans HC, Hoster E, Hermine O, et al.: Treatment of Older Patients With Mantle Cell Lymphoma (MCL): Long-Term Follow-Up of the Randomized European MCL Elderly Trial. J Clin Oncol 38 (3): 248-256, 2020.
- Rummel MJ, Niederle N, Maschmeyer G, et al.: Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 381 (9873): 1203-10, 2013.
- Robak T, Jin J, Pylypenko H, et al.: Frontline bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) versus rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in transplantation-ineligible patients with newly diagnosed mantle cell lymphoma: final overall survival results of a randomised, open-label, phase 3 study. Lancet Oncol 19 (11): 1449-1458, 2018.
- Hermine O, Hoster E, Walewski J, et al.: Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet 388 (10044): 565-75, 2016.
- Hermine O, Jiang L, Walewski J, et al.: Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL younger): a long-term follow-up of the randomized, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. [Abstract] Blood 138 (Suppl 1); A-380, 2021.
- Hermine O, Jiang L, Walewski J, et al.: High-Dose Cytarabine and Autologous Stem-Cell Transplantation in Mantle Cell Lymphoma: Long-Term Follow-Up of the Randomized Mantle Cell Lymphoma Younger Trial of the European Mantle Cell Lymphoma Network. J Clin Oncol 41 (3): 479-484, 2023.
- Khouri IF, Lee MS, Saliba RM, et al.: Nonablative allogeneic stem-cell transplantation for advanced/recurrent mantle-cell lymphoma. J Clin Oncol 21 (23): 4407-12, 2003.
- Dreyling M, Lenz G, Hoster E, et al.: Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood 105 (7): 2677-84, 2005.
- Geisler CH, Kolstad A, Laurell A, et al.: Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 112 (7): 2687-93, 2008.
- Tam CS, Bassett R, Ledesma C, et al.: Mature results of the M. D. Anderson Cancer Center risk-adapted transplantation strategy in mantle cell lymphoma. Blood 113 (18): 4144-52, 2009.
- Damon LE, Johnson JL, Niedzwiecki D, et al.: Immunochemotherapy and autologous stem-cell transplantation for untreated patients with mantle-cell lymphoma: CALGB 59909. J Clin Oncol 27 (36): 6101-8, 2009.
- Fenske TS, Zhang MJ, Carreras J, et al.: Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 32 (4): 273-81, 2014.
- Martin P, Cohen JB, Wang M, et al.: Treatment Outcomes and Roles of Transplantation and Maintenance Rituximab in Patients With Previously Untreated Mantle Cell Lymphoma: Results From Large Real-World Cohorts. J Clin Oncol 41 (3): 541-554, 2023.
- Le Gouill S, Thieblemont C, Oberic L, et al.: Rituximab after Autologous Stem-Cell Transplantation in Mantle-Cell Lymphoma. N Engl J Med 377 (13): 1250-1260, 2017.
- Wang M, Fayad L, Wagner-Bartak N, et al.: Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol 13 (7): 716-23, 2012.
- Trněný M, Lamy T, Walewski J, et al.: Lenalidomide versus investigator's choice in relapsed or refractory mantle cell lymphoma (MCL-002; SPRINT): a phase 2, randomised, multicentre trial. Lancet Oncol 17 (3): 319-31, 2016.
- Wang M, Rule S, Zinzani PL, et al.: Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet 391 (10121): 659-667, 2018.
- Song Y, Zhou K, Zou D, et al.: Zanubrutinib in relapsed/refractory mantle cell lymphoma: long-term efficacy and safety results from a phase 2 study. Blood 139 (21): 3148-3158, 2022.
- Wang M, Munoz J, Goy A, et al.: Three-Year Follow-Up of KTE-X19 in Patients With Relapsed/Refractory Mantle Cell Lymphoma, Including High-Risk Subgroups, in the ZUMA-2 Study. J Clin Oncol 41 (3): 555-567, 2023.
- Wang Y, Jain P, Locke FL, et al.: Brexucabtagene Autoleucel for Relapsed or Refractory Mantle Cell Lymphoma in Standard-of-Care Practice: Results From the US Lymphoma CAR T Consortium. J Clin Oncol 41 (14): 2594-2606, 2023.
- Wang ML, Jurczak W, Zinzani PL, et al.: Pirtobrutinib in Covalent Bruton Tyrosine Kinase Inhibitor Pretreated Mantle-Cell Lymphoma. J Clin Oncol 41 (24): 3988-3997, 2023.
- Martin P, Ruan J, Leonard JP: The potential for chemotherapy-free strategies in mantle cell lymphoma. Blood 130 (17): 1881-1888, 2017.
- Forstpointner R, Unterhalt M, Dreyling M, et al.: Maintenance therapy with rituximab leads to a significant prolongation of response duration after salvage therapy with a combination of rituximab, fludarabine, cyclophosphamide, and mitoxantrone (R-FCM) in patients with recurring and refractory follicular and mantle cell lymphomas: Results of a prospective randomized study of the German Low Grade Lymphoma Study Group (GLSG). Blood 108 (13): 4003-8, 2006.
- Blum KA, Lozanski G, Byrd JC: Adult Burkitt leukemia and lymphoma. Blood 104 (10): 3009-20, 2004.
- Onciu M, Schlette E, Zhou Y, et al.: Secondary chromosomal abnormalities predict outcome in pediatric and adult high-stage Burkitt lymphoma. Cancer 107 (5): 1084-92, 2006.
- Macpherson N, Lesack D, Klasa R, et al.: Small noncleaved, non-Burkitt's (Burkit-Like) lymphoma: cytogenetics predict outcome and reflect clinical presentation. J Clin Oncol 17 (5): 1558-67, 1999.
- Dave SS, Fu K, Wright GW, et al.: Molecular diagnosis of Burkitt's lymphoma. N Engl J Med 354 (23): 2431-42, 2006.
- Hummel M, Bentink S, Berger H, et al.: A biologic definition of Burkitt's lymphoma from transcriptional and genomic profiling. N Engl J Med 354 (23): 2419-30, 2006.
- Salaverria I, Siebert R: The gray zone between Burkitt's lymphoma and diffuse large B-cell lymphoma from a genetics perspective. J Clin Oncol 29 (14): 1835-43, 2011.
- Thomas DA, Faderl S, O'Brien S, et al.: Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer 106 (7): 1569-80, 2006.
- Dunleavy K, Pittaluga S, Shovlin M, et al.: Low-intensity therapy in adults with Burkitt's lymphoma. N Engl J Med 369 (20): 1915-25, 2013.
- Hoelzer D, Walewski J, Döhner H, et al.: Improved outcome of adult Burkitt lymphoma/leukemia with rituximab and chemotherapy: report of a large prospective multicenter trial. Blood 124 (26): 3870-9, 2014.
- Ribrag V, Koscielny S, Bosq J, et al.: Rituximab and dose-dense chemotherapy for adults with Burkitt's lymphoma: a randomised, controlled, open-label, phase 3 trial. Lancet 387 (10036): 2402-11, 2016.
- Magrath I, Adde M, Shad A, et al.: Adults and children with small non-cleaved-cell lymphoma have a similar excellent outcome when treated with the same chemotherapy regimen. J Clin Oncol 14 (3): 925-34, 1996.
- Hoelzer D, Ludwig WD, Thiel E, et al.: Improved outcome in adult B-cell acute lymphoblastic leukemia. Blood 87 (2): 495-508, 1996.
- Lee EJ, Petroni GR, Schiffer CA, et al.: Brief-duration high-intensity chemotherapy for patients with small noncleaved-cell lymphoma or FAB L3 acute lymphocytic leukemia: results of cancer and leukemia group B study 9251. J Clin Oncol 19 (20): 4014-22, 2001.
- Mead GM, Sydes MR, Walewski J, et al.: An international evaluation of CODOX-M and CODOX-M alternating with IVAC in adult Burkitt's lymphoma: results of United Kingdom Lymphoma Group LY06 study. Ann Oncol 13 (8): 1264-74, 2002.
- Rizzieri DA, Johnson JL, Niedzwiecki D, et al.: Intensive chemotherapy with and without cranial radiation for Burkitt leukemia and lymphoma: final results of Cancer and Leukemia Group B Study 9251. Cancer 100 (7): 1438-48, 2004.
- Noy A, Lee JY, Cesarman E, et al.: AMC 048: modified CODOX-M/IVAC-rituximab is safe and effective for HIV-associated Burkitt lymphoma. Blood 126 (2): 160-6, 2015.
- Verdonck LF, Dekker AW, de Gast GC, et al.: Autologous bone marrow transplantation for adult poor-risk lymphoblastic lymphoma in first remission. J Clin Oncol 10 (4): 644-6, 1992.
- Thomas DA, O'Brien S, Cortes J, et al.: Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood 104 (6): 1624-30, 2004.
- Sweetenham JW, Santini G, Qian W, et al.: High-dose therapy and autologous stem-cell transplantation versus conventional-dose consolidation/maintenance therapy as postremission therapy for adult patients with lymphoblastic lymphoma: results of a randomized trial of the European Group for Blood and Marrow Transplantation and the United Kingdom Lymphoma Group. J Clin Oncol 19 (11): 2927-36, 2001.
- Nador RG, Cesarman E, Chadburn A, et al.: Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpes virus. Blood 88 (2): 645-56, 1996.
- Shimada K, Hayakawa F, Kiyoi H: Biology and management of primary effusion lymphoma. Blood 132 (18): 1879-1888, 2018.
- Castillo JJ, Bibas M, Miranda RN: The biology and treatment of plasmablastic lymphoma. Blood 125 (15): 2323-30, 2015.
- Al-Malki MM, Castillo JJ, Sloan JM, et al.: Hematopoietic cell transplantation for plasmablastic lymphoma: a review. Biol Blood Marrow Transplant 20 (12): 1877-84, 2014.
- Cattaneo C, Re A, Ungari M, et al.: Plasmablastic lymphoma among human immunodeficiency virus-positive patients: results of a single center's experience. Leuk Lymphoma 56 (1): 267-9, 2015.
- Morrison VA, Dunn DL, Manivel JC, et al.: Clinical characteristics of post-transplant lymphoproliferative disorders. Am J Med 97 (1): 14-24, 1994.
- Knowles DM, Cesarman E, Chadburn A, et al.: Correlative morphologic and molecular genetic analysis demonstrates three distinct categories of posttransplantation lymphoproliferative disorders. Blood 85 (2): 552-65, 1995.
- Leblond V, Dhedin N, Mamzer Bruneel MF, et al.: Identification of prognostic factors in 61 patients with posttransplantation lymphoproliferative disorders. J Clin Oncol 19 (3): 772-8, 2001.
- Ghobrial IM, Habermann TM, Maurer MJ, et al.: Prognostic analysis for survival in adult solid organ transplant recipients with post-transplantation lymphoproliferative disorders. J Clin Oncol 23 (30): 7574-82, 2005.
- Evens AM, David KA, Helenowski I, et al.: Multicenter analysis of 80 solid organ transplantation recipients with post-transplantation lymphoproliferative disease: outcomes and prognostic factors in the modern era. J Clin Oncol 28 (6): 1038-46, 2010.
- Dierickx D, Tousseyn T, Gheysens O: How I treat posttransplant lymphoproliferative disorders. Blood 126 (20): 2274-83, 2015.
- Kuehnle I, Huls MH, Liu Z, et al.: CD20 monoclonal antibody (rituximab) for therapy of Epstein-Barr virus lymphoma after hemopoietic stem-cell transplantation. Blood 95 (4): 1502-5, 2000.
- Trappe RU, Dierickx D, Zimmermann H, et al.: Response to Rituximab Induction Is a Predictive Marker in B-Cell Post-Transplant Lymphoproliferative Disorder and Allows Successful Stratification Into Rituximab or R-CHOP Consolidation in an International, Prospective, Multicenter Phase II Trial. J Clin Oncol 35 (5): 536-543, 2017.
- Leblond V, Sutton L, Dorent R, et al.: Lymphoproliferative disorders after organ transplantation: a report of 24 cases observed in a single center. J Clin Oncol 13 (4): 961-8, 1995.
- Mamzer-Bruneel MF, Lomé C, Morelon E, et al.: Durable remission after aggressive chemotherapy for very late post-kidney transplant lymphoproliferation: A report of 16 cases observed in a single center. J Clin Oncol 18 (21): 3622-32, 2000.
- Swinnen LJ: Durable remission after aggressive chemotherapy for post-cardiac transplant lymphoproliferation. Leuk Lymphoma 28 (1-2): 89-101, 1997.
- McCarthy M, Ramage J, McNair A, et al.: The clinical diversity and role of chemotherapy in lymphoproliferative disorder in liver transplant recipients. J Hepatol 27 (6): 1015-21, 1997.
- Leblond V, Davi F, Charlotte F, et al.: Posttransplant lymphoproliferative disorders not associated with Epstein-Barr virus: a distinct entity? J Clin Oncol 16 (6): 2052-9, 1998.
- Senderowicz AM, Vitetta E, Headlee D, et al.: Complete sustained response of a refractory, post-transplantation, large B-cell lymphoma to an anti-CD22 immunotoxin. Ann Intern Med 126 (11): 882-5, 1997.
- Haddad E, Paczesny S, Leblond V, et al.: Treatment of B-lymphoproliferative disorder with a monoclonal anti-interleukin-6 antibody in 12 patients: a multicenter phase 1-2 clinical trial. Blood 97 (6): 1590-7, 2001.
- Guinee D, Jaffe E, Kingma D, et al.: Pulmonary lymphomatoid granulomatosis. Evidence for a proliferation of Epstein-Barr virus infected B-lymphocytes with a prominent T-cell component and vasculitis. Am J Surg Pathol 18 (8): 753-64, 1994.
- Myers JL, Kurtin PJ, Katzenstein AL, et al.: Lymphomatoid granulomatosis. Evidence of immunophenotypic diversity and relationship to Epstein-Barr virus infection. Am J Surg Pathol 19 (11): 1300-12, 1995.
Otros trastornos linfoproliferativos y relacionados
Enfermedad de Castleman
Es posible que el resultado de una biopsia de las acumulaciones localizadas o multifocales de los ganglios linfáticos conduzca a un diagnóstico de la enfermedad de Castleman (EC), aunque es un diagnóstico poco frecuente. En sentido estricto, no se trata de un linfoma, ni siquiera de una neoplasia maligna. Sin embargo, hematólogos u oncólogos atienden y tratan a muchos pacientes con EC.
La EC localizada o unicéntrica suele ser asintomática y se produce en el mediastino, que es la presentación más común de la EC. Para esta forma de crecimiento lento se utiliza la observación cautelosa, la cirugía o la radioterapia. La EC multicéntrica (30 % de los pacientes con EC) se presenta con linfadenopatía en múltiples sitios; síntomas como fiebre, sudoración nocturna, pérdida de peso y fatiga; y anomalías de las pruebas de laboratorio como anemia, albúmina baja, proteína C-reactiva elevada y fibrinógeno alto. La EC multicéntrica (ECM) se subdivide en ECM relacionada con el virus del herpes humano 8 (normalmente con VIH o con inmunodeficiencia grave) o ECM idiopática. Las citopenias y la hipercitocinemia se atribuyen a la sobreproducción de interleucina-6 (IL-6). La ECM es una característica observada en el síndrome POEMS (polineuropatía, organomegalia, endocrinopatía, gammapatía monoclonal y anomalías cutáneas) y el síndrome TAFRO (trombocitopenia, anasarca, fiebre, reticulinofibrosis y organomegalia). El tratamiento con siltuximab (un anticuerpo monoclonal anti–IL-6), rituximab (un anticuerpo monoclonal anti-CD20) o quimioterapéuticos se ha notificado en series anecdóticas no aleatorias.
Linfoma histiocítico verdadero
Los linfomas histiocíticos verdaderos producen tumores muy poco frecuentes que exhiben diferenciación histiocítica y expresan marcadores histiocíticos en ausencia de marcadores inmunitarios de linaje específico de células o linfocitos B o T. También se llaman sarcomas histiocíticos o linfomas histiocitarios verdaderos. Se debe tener cuidado con las pruebas inmunofenotípicas para excluir el LACG o los síndromes hemofagocíticos que se presentan debido a infecciones víricas, en especial las causadas por el virus de Epstein-Barr.
Opciones de tratamiento
El tratamiento es similar al de los linfomas difusos de células grandes en estadio comparable, pero todavía queda por definir el abordaje óptimo.
References
- van Rhee F, Voorhees P, Dispenzieri A, et al.: International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 132 (20): 2115-2124, 2018.
- Dispenzieri A: POEMS Syndrome: 2019 Update on diagnosis, risk-stratification, and management. Am J Hematol 94 (7): 812-827, 2019.
- Zhang Y, Suo SS, Yang HJ, et al.: Clinical features and treatment of 7 Chinese TAFRO syndromes from 96 de novo Castleman diseases: a 10-year retrospective study. J Cancer Res Clin Oncol 146 (2): 357-365, 2020.
- Fujimoto S, Sakai T, Kawabata H, et al.: Is TAFRO syndrome a subtype of idiopathic multicentric Castleman disease? Am J Hematol 94 (9): 975-983, 2019.
- Tonialini L, Bonfichi M, Ferrero S, et al.: Siltuximab in relapsed/refractory multicentric Castleman disease: Experience of the Italian NPP program. Hematol Oncol 36 (4): 689-692, 2018.
- Dong Y, Zhang L, Nong L, et al.: Effectiveness of rituximab-containing treatment regimens in idiopathic multicentric Castleman disease. Ann Hematol 97 (9): 1641-1647, 2018.
- Zhang L, Zhao AL, Duan MH, et al.: Phase 2 study using oral thalidomide-cyclophosphamide-prednisone for idiopathic multicentric Castleman disease. Blood 133 (16): 1720-1728, 2019.
- van Rhee F, Wong RS, Munshi N, et al.: Siltuximab for multicentric Castleman's disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol 15 (9): 966-74, 2014.
- Soslow RA, Davis RE, Warnke RA, et al.: True histiocytic lymphoma following therapy for lymphoblastic neoplasms. Blood 87 (12): 5207-12, 1996.
- Kamel OW, Gocke CD, Kell DL, et al.: True histiocytic lymphoma: a study of 12 cases based on current definition. Leuk Lymphoma 18 (1-2): 81-6, 1995.
Aspectos generales de las opciones de tratamiento del linfoma no Hodgkin de células B
El tratamiento del linfoma no Hodgkin (LNH) depende del tipo histológico y el estadio. Muchas de las mejoras en la supervivencia se han logrado mediante ensayos clínicos (terapia experimental) en los que se intentó perfeccionar el mejor tratamiento aceptado disponible (tratamiento de referencia o estándar).
En los pacientes asintomáticos con formas de LNH de crecimiento lento en estadio avanzado, es posible diferir el tratamiento hasta que el paciente presente síntomas o la enfermedad progrese. Cuando se difiere el tratamiento, la evolución clínica de los pacientes con LNH de crecimiento lento es variable; se necesita una observación frecuente y cuidadosa para poder iniciar un tratamiento eficaz cuando se acelera la evolución clínica de la enfermedad. Algunos pacientes tienen un período prolongado de enfermedad de crecimiento lento, pero en otros la enfermedad evoluciona rápido a tipos más malignos de LNH, lo que exige un tratamiento inmediato.
Las técnicas de radioterapia son un poco diferentes de las que se utilizan para el tratamiento del linfoma de Hodgkin. La dosis de radioterapia por lo habitual oscila entre 25 Gy y 50 Gy, y depende de factores, como el tipo histológico de linfoma, el estadio de la enfermedad, el estado general del paciente, el objetivo del tratamiento (curativo o paliativo), la proximidad a órganos circundantes sensibles, y si se usa radioterapia sola o combinada con quimioterapia. Debido a la variedad de presentaciones clínicas de la enfermedad y de la recaída, a veces se incluyen en el tratamiento sitios no habituales, como el anillo de Waldeyer, los ganglios epitrocleares o los ganglios mesentéricos. La morbilidad relacionada con el tratamiento se debe considerar en detalle. La mayoría de los pacientes que reciben radiación se tratan solo en un lado del diafragma. Las presentaciones localizadas del LNH extraganglionar se pueden tratar con técnicas dirigidas al campo comprometido con éxito significativo (>50 %).
| Estadio | Opciones de tratamiento |
|---|---|
| TMO = trasplante de médula ósea; CAR = receptor de antígeno quimérico; SNC = sistema nervioso central; RTCC = radioterapia de campo comprometido; PI3K = fosfatidilinositol-3–cinasa; R-ACVBP = rituximab, doxorrubicina, ciclofosfamida, vindesina, bleomicina y prednisona; R-CHOP = rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona; TCM = trasplante de células madre. | |
| Linfoma no Hodgkin de células B de crecimiento lento en estadio I y linfoma no Hodgkin de crecimiento lento y adyacente en estadio II | Radioterapia |
| Rituximab con quimioterapia o sin esta | |
| Observación cautelosa | |
| Otros tratamientos indicados para pacientes con enfermedad en estadio avanzado | |
| Linfoma no Hodgkin de células B de crecimiento lento no adyacente en estadios II, III y IV | Observación cautelosa para pacientes asintomáticos |
| Rituximab solo o en combinación con citotóxicos utilizados en el tratamiento de primera línea | |
| Lenalidomida y rituximab | |
| Mantenimiento con rituximab | |
| Obinutuzumab solo o en combinación con citotóxicos utilizados en el tratamiento de primera línea | |
| Linfoma no Hodgkin de células B de crecimiento lento recidivante | Rituximab solo o en combinación con citotóxicos utilizados en el tratamiento de primera línea |
| Obinutuzumab solo o en combinación con citotóxicos utilizados en el tratamiento de primera línea | |
| Lenalidomida y rituximab | |
| Inhibidor de EZH2 | |
| Captadores biespecíficos de células T | |
| Terapia de células T con CAR | |
| TCM | |
| Inhibidor de PI3K | |
| Radioterapia paliativa | |
| Linfoma no Hodgkin de células B de crecimiento rápido en estadio I y linfoma no Hodgkin de células B de crecimiento rápido adyacente en estadio II | R-CHOP con RTCC o sin esta |
| R-ACVBP (en evaluación clínica) | |
| Linfoma no Hodgkin de células B de crecimiento rápido no adyacente en estadios II, III o IV | Pola-R-CHP |
| R-CHOP | |
| Otra quimioterapia combinada | |
| Consolidación con radioterapia dirigida a los sitios de enfermedad con gran masa tumoral (en evaluación clínica) | |
| Linfoma no Hodgkin de células B de crecimiento rápido recidivante | Terapia de células T con CAR para la enfermedad primaria resistente al tratamiento o la recaída dentro de 1 año |
| Consolidación con TMO o TCM | |
| Terapia de células T con CAR para la recaída después de un TCM autógeno | |
| Tafasitamab y lenalidomida | |
| Rituximab y lenalidomida | |
| Polatuzumab vedotina, rituximab y bendamustina | |
| Loncastuximab tesirina | |
| Captadores biespecíficos de células T | |
| Radioterapia paliativa | |
| Linfoma linfoblástico de células B o leucemia linfocítica aguda | Tratamiento intensivo |
| Radioterapia | |
| Linfoma difuso de células pequeñas no hendidas o linfoma de Burkitt | Regímenes multifarmacológicos intensivos |
| Profilaxis del SNC | |
Aunque los tratamientos existentes son curativos en una proporción significativa de los pacientes con linfoma, se están llevando a cabo numerosos ensayos clínicos para mejorar el tratamiento. En lo posible, los pacientes se deben incluir en estos estudios. Se propusieron pautas estandarizadas para evaluar la respuesta en los ensayos clínicos.
En varias revisiones retrospectivas se indicó que la vigilancia rutinaria mediante imágenes aporta poco o ningún valor a los pacientes con linfoma difuso de células B grandes (LDCBG) que logran una remisión clínica completa después de la terapia de inducción. Además, resulta difícil determinar el valor pronóstico de la tomografía por emisión de positrones-tomografía computarizada provisional durante la terapia de inducción del LDCBG.
Los linfomas de crecimiento rápido se observan cada vez más en pacientes con VIH. El tratamiento de estos pacientes exige consideraciones especiales. Para obtener más información, consultar Tratamiento del linfoma relacionado con el SIDA
Además de los exámenes de detección del VIH en pacientes con linfomas de crecimiento rápido, se puede evaluar la presencia de hepatitis B o C activa antes del tratamiento con rituximab o quimioterapia. Los pacientes con infección por el virus de la hepatitis B (VHB) detectable se benefician de la profilaxis con entecavir en el entorno del tratamiento con rituximab. Los pacientes con infección por el VHB resuelta (es decir, negativa para el antígeno de superficie del VHB, pero positiva para el anticuerpo central del VHB), presentan riesgo de reactivación de la infección por el VHB y necesitan vigilancia del DNA del VHB. La terapia profiláctica con nucleósidos redujo la reactivación de la infección por el VHB de un 10,8 % a un 2,1 % en un estudio retrospectivo de 326 pacientes. De forma similar, los pacientes que reciben rituximab con quimioterapia combinada o sin esta suelen recibir profilaxis con aciclovir o valaciclovir para el virus del herpes zóster y profilaxis con trimetoprima-sulfametoxazol o dapsona para la infección por Pneumocystis. El deterioro inmunitario a largo plazo se evaluó en un estudio retrospectivo de cohorte que incluyó 21 690 sobrevivientes de LDCBG del California Cancer Registry. Se encontraron cocientes de tasas de incidencia elevadas hasta 10 años después de neumonía (10,8 veces mayor), meningitis (5,3 veces mayor), deficiencia de inmunoglobulina (17,6 veces mayor) y citopenias autoinmunitarias (12 veces mayor).
En un registro danés de 2508 pacientes, la incidencia de insuficiencia cardíaca congestiva inducida por doxorrubicina aumentó en 115 sobrevivientes del LNH con antecedentes de enfermedad cardíaca (cociente de riesgos instantáneos [CRI], 2,71; intervalo de confianza [IC] 95 %, 1,15−6,36) o múltiples factores de riesgo cardiovascular (CRI, 2,86; IC 95 %, 1,56−5,23).
Hay varias presentaciones clínicas poco habituales de linfoma que a menudo exigen abordajes modificados para la estadificación y el tratamiento. El lector puede remitirse a otras revisiones para obtener descripciones más detalladas de otras presentaciones extraganglionares en el aparato digestivo, la tiroides, el bazo, los testículos, los senos paranasales, los huesos, la órbita y la piel.
Para obtener más información, consultar Tratamiento del linfoma primario del sistema nervioso central.
References
- Cheson BD, Horning SJ, Coiffier B, et al.: Report of an international workshop to standardize response criteria for non-Hodgkin's lymphomas. NCI Sponsored International Working Group. J Clin Oncol 17 (4): 1244, 1999.
- Mamot C, Klingbiel D, Hitz F, et al.: Final Results of a Prospective Evaluation of the Predictive Value of Interim Positron Emission Tomography in Patients With Diffuse Large B-Cell Lymphoma Treated With R-CHOP-14 (SAKK 38/07). J Clin Oncol 33 (23): 2523-9, 2015.
- Thompson CA, Ghesquieres H, Maurer MJ, et al.: Utility of routine post-therapy surveillance imaging in diffuse large B-cell lymphoma. J Clin Oncol 32 (31): 3506-12, 2014.
- El-Galaly TC, Jakobsen LH, Hutchings M, et al.: Routine Imaging for Diffuse Large B-Cell Lymphoma in First Complete Remission Does Not Improve Post-Treatment Survival: A Danish-Swedish Population-Based Study. J Clin Oncol 33 (34): 3993-8, 2015.
- Huntington SF, Svoboda J, Doshi JA: Cost-effectiveness analysis of routine surveillance imaging of patients with diffuse large B-cell lymphoma in first remission. J Clin Oncol 33 (13): 1467-74, 2015.
- Niitsu N, Hagiwara Y, Tanae K, et al.: Prospective analysis of hepatitis B virus reactivation in patients with diffuse large B-cell lymphoma after rituximab combination chemotherapy. J Clin Oncol 28 (34): 5097-100, 2010.
- Dong HJ, Ni LN, Sheng GF, et al.: Risk of hepatitis B virus (HBV) reactivation in non-Hodgkin lymphoma patients receiving rituximab-chemotherapy: a meta-analysis. J Clin Virol 57 (3): 209-14, 2013.
- Huang YH, Hsiao LT, Hong YC, et al.: Randomized controlled trial of entecavir prophylaxis for rituximab-associated hepatitis B virus reactivation in patients with lymphoma and resolved hepatitis B. J Clin Oncol 31 (22): 2765-72, 2013.
- Li H, Zhang HM, Chen LF, et al.: Prophylactic lamivudine to improve the outcome of HBsAg-positive lymphoma patients during chemotherapy: a systematic review and meta-analysis. Clin Res Hepatol Gastroenterol 39 (1): 80-92, 2015.
- Kusumoto S, Arcaini L, Hong X, et al.: Risk of HBV reactivation in patients with B-cell lymphomas receiving obinutuzumab or rituximab immunochemotherapy. Blood 133 (2): 137-146, 2019.
- Shree T, Li Q, Glaser SL, et al.: Impaired Immune Health in Survivors of Diffuse Large B-Cell Lymphoma. J Clin Oncol 38 (15): 1664-1675, 2020.
- Salz T, Zabor EC, de Nully Brown P, et al.: Preexisting Cardiovascular Risk and Subsequent Heart Failure Among Non-Hodgkin Lymphoma Survivors. J Clin Oncol 35 (34): 3837-3843, 2017.
- Maor MH, Velasquez WS, Fuller LM, et al.: Stomach conservation in stages IE and IIE gastric non-Hodgkin's lymphoma. J Clin Oncol 8 (2): 266-71, 1990.
- Salles G, Herbrecht R, Tilly H, et al.: Aggressive primary gastrointestinal lymphomas: review of 91 patients treated with the LNH-84 regimen. A study of the Groupe d'Etude des Lymphomes Agressifs. Am J Med 90 (1): 77-84, 1991.
- Taal BG, Burgers JM, van Heerde P, et al.: The clinical spectrum and treatment of primary non-Hodgkin's lymphoma of the stomach. Ann Oncol 4 (10): 839-46, 1993.
- Tondini C, Giardini R, Bozzetti F, et al.: Combined modality treatment for primary gastrointestinal non-Hodgkin's lymphoma: the Milan Cancer Institute experience. Ann Oncol 4 (10): 831-7, 1993.
- d'Amore F, Brincker H, Grønbaek K, et al.: Non-Hodgkin's lymphoma of the gastrointestinal tract: a population-based analysis of incidence, geographic distribution, clinicopathologic presentation features, and prognosis. Danish Lymphoma Study Group. J Clin Oncol 12 (8): 1673-84, 1994.
- Haim N, Leviov M, Ben-Arieh Y, et al.: Intermediate and high-grade gastric non-Hodgkin's lymphoma: a prospective study of non-surgical treatment with primary chemotherapy, with or without radiotherapy. Leuk Lymphoma 17 (3-4): 321-6, 1995.
- Koch P, del Valle F, Berdel WE, et al.: Primary gastrointestinal non-Hodgkin's lymphoma: I. Anatomic and histologic distribution, clinical features, and survival data of 371 patients registered in the German Multicenter Study GIT NHL 01/92. J Clin Oncol 19 (18): 3861-73, 2001.
- Koch P, del Valle F, Berdel WE, et al.: Primary gastrointestinal non-Hodgkin's lymphoma: II. Combined surgical and conservative or conservative management only in localized gastric lymphoma--results of the prospective German Multicenter Study GIT NHL 01/92. J Clin Oncol 19 (18): 3874-83, 2001.
- Koch P, Probst A, Berdel WE, et al.: Treatment results in localized primary gastric lymphoma: data of patients registered within the German multicenter study (GIT NHL 02/96). J Clin Oncol 23 (28): 7050-9, 2005.
- Blair TJ, Evans RG, Buskirk SJ, et al.: Radiotherapeutic management of primary thyroid lymphoma. Int J Radiat Oncol Biol Phys 11 (2): 365-70, 1985.
- Junor EJ, Paul J, Reed NS: Primary non-Hodgkin's lymphoma of the thyroid. Eur J Surg Oncol 18 (4): 313-21, 1992.
- Morel P, Dupriez B, Gosselin B, et al.: Role of early splenectomy in malignant lymphomas with prominent splenic involvement (primary lymphomas of the spleen). A study of 59 cases. Cancer 71 (1): 207-15, 1993.
- Zucca E, Conconi A, Mughal TI, et al.: Patterns of outcome and prognostic factors in primary large-cell lymphoma of the testis in a survey by the International Extranodal Lymphoma Study Group. J Clin Oncol 21 (1): 20-7, 2003.
- Vitolo U, Chiappella A, Ferreri AJ, et al.: First-line treatment for primary testicular diffuse large B-cell lymphoma with rituximab-CHOP, CNS prophylaxis, and contralateral testis irradiation: final results of an international phase II trial. J Clin Oncol 29 (20): 2766-72, 2011.
- Cheah CY, Wirth A, Seymour JF: Primary testicular lymphoma. Blood 123 (4): 486-93, 2014.
- Liang R, Todd D, Chan TK, et al.: Treatment outcome and prognostic factors for primary nasal lymphoma. J Clin Oncol 13 (3): 666-70, 1995.
- Cheung MM, Chan JK, Lau WH, et al.: Primary non-Hodgkin's lymphoma of the nose and nasopharynx: clinical features, tumor immunophenotype, and treatment outcome in 113 patients. J Clin Oncol 16 (1): 70-7, 1998.
- Hausdorff J, Davis E, Long G, et al.: Non-Hodgkin's lymphoma of the paranasal sinuses: clinical and pathological features, and response to combined-modality therapy. Cancer J Sci Am 3 (5): 303-11, 1997 Sep-Oct.
- Sasai K, Yamabe H, Kokubo M, et al.: Head-and-neck stages I and II extranodal non-Hodgkin's lymphomas: real classification and selection for treatment modality. Int J Radiat Oncol Biol Phys 48 (1): 153-60, 2000.
- Ferreri AJ, Reni M, Ceresoli GL, et al.: Therapeutic management with adriamycin-containing chemotherapy and radiotherapy of monostotic and polyostotic primary non-Hodgkin's lymphoma of bone in adults. Cancer Invest 16 (8): 554-61, 1998.
- Dubey P, Ha CS, Besa PC, et al.: Localized primary malignant lymphoma of bone. Int J Radiat Oncol Biol Phys 37 (5): 1087-93, 1997.
- Martinet S, Ozsahin M, Belkacémi Y, et al.: Outcome and prognostic factors in orbital lymphoma: a Rare Cancer Network study on 90 consecutive patients treated with radiotherapy. Int J Radiat Oncol Biol Phys 55 (4): 892-8, 2003.
- Uno T, Isobe K, Shikama N, et al.: Radiotherapy for extranodal, marginal zone, B-cell lymphoma of mucosa-associated lymphoid tissue originating in the ocular adnexa: a multiinstitutional, retrospective review of 50 patients. Cancer 98 (4): 865-71, 2003.
- Sjö LD, Ralfkiaer E, Juhl BR, et al.: Primary lymphoma of the lacrimal sac: an EORTC ophthalmic oncology task force study. Br J Ophthalmol 90 (8): 1004-9, 2006.
- Stefanovic A, Lossos IS: Extranodal marginal zone lymphoma of the ocular adnexa. Blood 114 (3): 501-10, 2009.
- Sjö LD: Ophthalmic lymphoma: epidemiology and pathogenesis. Acta Ophthalmol 87 Thesis 1: 1-20, 2009.
- Geelen FA, Vermeer MH, Meijer CJ, et al.: bcl-2 protein expression in primary cutaneous large B-cell lymphoma is site-related. J Clin Oncol 16 (6): 2080-5, 1998.
- Pandolfino TL, Siegel RS, Kuzel TM, et al.: Primary cutaneous B-cell lymphoma: review and current concepts. J Clin Oncol 18 (10): 2152-68, 2000.
- Sarris AH, Braunschweig I, Medeiros LJ, et al.: Primary cutaneous non-Hodgkin's lymphoma of Ann Arbor stage I: preferential cutaneous relapses but high cure rate with doxorubicin-based therapy. J Clin Oncol 19 (2): 398-405, 2001.
- Grange F, Bekkenk MW, Wechsler J, et al.: Prognostic factors in primary cutaneous large B-cell lymphomas: a European multicenter study. J Clin Oncol 19 (16): 3602-10, 2001.
- Mirza I, Macpherson N, Paproski S, et al.: Primary cutaneous follicular lymphoma: an assessment of clinical, histopathologic, immunophenotypic, and molecular features. J Clin Oncol 20 (3): 647-55, 2002.
- Smith BD, Glusac EJ, McNiff JM, et al.: Primary cutaneous B-cell lymphoma treated with radiotherapy: a comparison of the European Organization for Research and Treatment of Cancer and the WHO classification systems. J Clin Oncol 22 (4): 634-9, 2004.
- Willemze R, Jaffe ES, Burg G, et al.: WHO-EORTC classification for cutaneous lymphomas. Blood 105 (10): 3768-85, 2005.
- El-Helw L, Goodwin S, Slater D, et al.: Primary B-cell lymphoma of the skin: the Sheffield Lymphoma Group Experience (1984-2003). Int J Oncol 25 (5): 1453-8, 2004.
- Zinzani PL, Quaglino P, Pimpinelli N, et al.: Prognostic factors in primary cutaneous B-cell lymphoma: the Italian Study Group for Cutaneous Lymphomas. J Clin Oncol 24 (9): 1376-82, 2006.
- Senff NJ, Noordijk EM, Kim YH, et al.: European Organization for Research and Treatment of Cancer and International Society for Cutaneous Lymphoma consensus recommendations for the management of cutaneous B-cell lymphomas. Blood 112 (5): 1600-9, 2008.
Tratamiento del linfoma no Hodgkin de células B de crecimiento lento en estadio I y del linfoma no Hodgkin de células B de crecimiento lento adyacente en estadio II
Aunque es poco común observar presentaciones localizadas del linfoma no Hodgkin (LNH) de células B, el objetivo del tratamiento es curarlo en los pacientes en los que se confirme la enfermedad localizada después de la estadificación apropiada.
Opciones de tratamiento del linfoma no Hodgkin de células B de crecimiento lento en estadio I y el linfoma no Hodgkin de células B de crecimiento lento adyacente en estadio II
Las opciones de tratamiento del linfoma no Hodgkin de células B de crecimiento lento en estadio I y el linfoma no Hodgkin de células B de crecimiento lento adyacente en estadio II son las siguientes:
- Radioterapia.
- Rituximab con quimioterapia o sin esta.
- Observación cautelosa.
- Otros tratamientos, como los diseñados para pacientes con enfermedad en estadio avanzado.
En un ensayo prospectivo aleatorizado, 150 pacientes de linfoma folicular en estadio I o estadio II se asignaron en forma aleatoria a recibir 30 Gy de radioterapia de campo comprometido sola o radioterapia y 6 ciclos de R-CVP (rituximab, ciclofosfamida, vincristina y prednisona). Después de una mediana de seguimiento de 9,6 años, la tasa de supervivencia sin progresión (SSP) a 10 años favoreció la terapia de modalidad combinada en un 59 % (intervalo de confianza [IC] 95 %, 46–74 %) versus un 41 % para la radioterapia sola (IC 95 %, 30–57 %) (P = 0,033). No hubo diferencia en la supervivencia general (SG) (87 y 95 %, P = 0,40).[Nivel de evidencia B1]
En el National Lymphocare Study se identificaron 471 pacientes con linfoma folicular en estadio I. De estos pacientes, 206 se estadificaron de manera cuidadosa mediante aspirado y biopsia de médula ósea, tomografía computarizada (TC) o tomografía por emisión de positrones con tomografía computarizada (TEP-TC). Los tratamientos no aleatorizados incluyeron radioterapia (27 %), quimioterapia con rituximab (quimioterapia R) (28 %), observación cautelosa (17 %), quimioterapia R con radioterapia (13 %) y rituximab solo (12 %), aunque más de un tercio de los pacientes comenzó el estudio en el grupo de terapia diferida. Al cabo de una mediana de seguimiento de 57 meses, la SSP favoreció la quimioterapia R o la quimioterapia R con radioterapia, pero la SG fue casi idéntica, superior al 90 % para todos los pacientes.[Nivel de evidencia C2] Se necesitan ensayos clínicos para responder a preguntas como las siguientes:
- ¿Se prefiere la observación cautelosa o la radioterapia después de una TEP-TC normal tras la biopsia por escisión?
- ¿Se debería agregar rituximab a la radioterapia para el linfoma folicular en estadio I?
- ¿La quimioterapia R con radioterapia cumple alguna función en el tratamiento?
Radioterapia
Se puede lograr el control a largo plazo de la enfermedad en los campos de radiación en un número significativo de pacientes con LNH de crecimiento lento en estadio I o estadio II mediante el uso de dosificaciones de radiación que oscilan entre 25 Gy y 40 Gy dirigidas a los sitios comprometidos o a los campos extendidos que cubren sitios ganglionares adyacentes. Casi la mitad de todos los pacientes tratados con radioterapia sola tendrán recaída fuera del campo de radiación en un plazo de 10 años.
En una revisión retrospectiva de 512 pacientes de un consorcio internacional, se evaluó a pacientes con linfoma folicular en estadio temprano que habían recibido al menos 24 Gy de radioterapia localizada en el momento de la presentación inicial. Al cabo de una mediana de seguimiento de 52 meses, 29,1 % de los pacientes presentaron linfoma recidivante en una mediana de 23 meses (intervalo, 1−143 meses). Al cabo de una mediana de seguimiento de 33 meses después de la recidiva, la tasa de SG a 3 años fue del 91,4 % una vez que los pacientes recibieron quimioterapia sistémica subsiguiente que incluyó rituximab, por lo general con quimioterapia.
El uso de radioterapia de dosis muy bajas de 4 Gy (2 Gy en 2 fracciones) a veces conlleva tasas de remisión en el 50 % de los pacientes que no toleran dosis más altas. En un ensayo prospectivo aleatorizado multicéntrico 548 pacientes con linfoma de zona marginal o linfoma folicular recibieron radioterapia: 4 Gy en 2 fracciones o 24 Gy en 12 fracciones.
- Al cabo de una mediana de seguimiento de 73,8 meses, la tasa de respuesta completa local a 5 años fue del 89,9 % (85,5–93,1 %) después de 24 Gy y del 70,4 % (64,7–75,4 %) después de 4 Gy (cociente de riesgos instantáneos [CRI], 3,46; IC 95 %, 2,25–5,33, P < 0,0001).
- Aunque el control local duradero fue superior en los pacientes que recibieron 24 Gy, el régimen de 4 Gy fue casi similar con una exposición a la radiación, una duración del tratamiento y un costo inferiores.
Es posible considerar el uso de terapia con protones para reducir la dosis de radiación dirigida a los órganos en riesgo, cuando la radiación mediastínica abarca el lado izquierdo del corazón o se sabe que aumentará el riesgo de cáncer de mama en mujeres jóvenes.
Rituximab con quimioterapia o sin esta
Cuando se contraindica la radioterapia para los pacientes sintomáticos que necesitan tratamiento, o cuando se prefiere un tratamiento alternativo, se puede administrar rituximab con quimioterapia o sin esta (como se indica para los pacientes en estadios más avanzados en la sección Rituximab solo o en combinación con fármacos citotóxicos utilizados en el tratamiento de primera línea). La utilidad del tratamiento adyuvante para disminuir la recaída mediante radioterapia y rituximab (un anticuerpo monoclonal anti-CD20), solo o en combinación con quimioterapia, se extrapoló de ensayos con pacientes de enfermedad en estadio avanzado y no se ha confirmado.
Observación cautelosa
Se puede considerar la observación cautelosa para pacientes asintomáticos. La observación cautelosa nunca se ha comparado con la radioterapia inicial en ningún ensayo prospectivo aleatorizado. En un análisis retrospectivo de la base de datos del Surveillance, Epidemiology and End Results (SEER) Program de pacientes diagnosticados a lo largo de 30 años, se observaron mejores desenlaces con la administración de radioterapia inicial.
Otros tratamientos indicados para pacientes con enfermedad en estadio avanzado
Los pacientes con enfermedad que no es posible tratar mediante radioterapia reciben el mismo tratamiento de los pacientes con linfoma de grado bajo en estadio III o en estadio IV.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
References
- MacManus M, Fisher R, Roos D, et al.: Randomized Trial of Systemic Therapy After Involved-Field Radiotherapy in Patients With Early-Stage Follicular Lymphoma: TROG 99.03. J Clin Oncol 36 (29): 2918-2925, 2018.
- Friedberg JW, Byrtek M, Link BK, et al.: Effectiveness of first-line management strategies for stage I follicular lymphoma: analysis of the National LymphoCare Study. J Clin Oncol 30 (27): 3368-75, 2012.
- Montoto S: Management of localized-stage follicular lymphoma: changing the paradigm? J Clin Oncol 30 (27): 3328-9, 2012.
- Haas RL, Poortmans P, de Jong D, et al.: High response rates and lasting remissions after low-dose involved field radiotherapy in indolent lymphomas. J Clin Oncol 21 (13): 2474-80, 2003.
- Guckenberger M, Alexandrow N, Flentje M: Radiotherapy alone for stage I-III low grade follicular lymphoma: long-term outcome and comparison of extended field and total nodal irradiation. Radiat Oncol 7: 103, 2012.
- Brady JL, Binkley MS, Hajj C, et al.: Definitive radiotherapy for localized follicular lymphoma staged by 18F-FDG PET-CT: a collaborative study by ILROG. Blood 133 (3): 237-245, 2019.
- Guadagnolo BA, Li S, Neuberg D, et al.: Long-term outcome and mortality trends in early-stage, Grade 1-2 follicular lymphoma treated with radiation therapy. Int J Radiat Oncol Biol Phys 64 (3): 928-34, 2006.
- Binkley MS, Brady JL, Hajj C, et al.: Salvage Treatment and Survival for Relapsed Follicular Lymphoma Following Primary Radiation Therapy: A Collaborative Study on Behalf of ILROG. Int J Radiat Oncol Biol Phys 104 (3): 522-529, 2019.
- Hoskin PJ, Kirkwood AA, Popova B, et al.: 4 Gy versus 24 Gy radiotherapy for patients with indolent lymphoma (FORT): a randomised phase 3 non-inferiority trial. Lancet Oncol 15 (4): 457-63, 2014.
- Hoskin P, Popova B, Schofield O, et al.: 4 Gy versus 24 Gy radiotherapy for follicular and marginal zone lymphoma (FoRT): long-term follow-up of a multicentre, randomised, phase 3, non-inferiority trial. Lancet Oncol 22 (3): 332-340, 2021.
- Dabaja BS, Hoppe BS, Plastaras JP, et al.: Proton therapy for adults with mediastinal lymphomas: the International Lymphoma Radiation Oncology Group guidelines. Blood 132 (16): 1635-1646, 2018.
- Cartron G, Bachy E, Tilly H, et al.: Randomized Phase III Trial Evaluating Subcutaneous Rituximab for the First-Line Treatment of Low-Tumor Burden Follicular Lymphoma: Results of a LYSA Study. J Clin Oncol 41 (19): 3523-3533, 2023.
- Kelsey SM, Newland AC, Hudson GV, et al.: A British National Lymphoma Investigation randomised trial of single agent chlorambucil plus radiotherapy versus radiotherapy alone in low grade, localised non-Hodgkins lymphoma. Med Oncol 11 (1): 19-25, 1994.
- Seymour JF, Pro B, Fuller LM, et al.: Long-term follow-up of a prospective study of combined modality therapy for stage I-II indolent non-Hodgkin's lymphoma. J Clin Oncol 21 (11): 2115-22, 2003.
- Advani R, Rosenberg SA, Horning SJ: Stage I and II follicular non-Hodgkin's lymphoma: long-term follow-up of no initial therapy. J Clin Oncol 22 (8): 1454-9, 2004.
- Pugh TJ, Ballonoff A, Newman F, et al.: Improved survival in patients with early stage low-grade follicular lymphoma treated with radiation: a Surveillance, Epidemiology, and End Results database analysis. Cancer 116 (16): 3843-51, 2010.
Tratamiento del linfoma no Hodgkin de células B de crecimiento lento no adyacente en estadio II, III o IV
El tratamiento óptimo para el linfoma no Hodgkin (LNH) de grado bajo en estadios avanzados es polémico debido a las tasas bajas de curación que se obtienen con las opciones terapéuticas vigentes. Hay numerosos ensayos clínicos en curso para evaluar asuntos relacionados con el tratamiento y se insta a los pacientes a que participen en estos. La tasa de recaída es muy constante a lo largo del tiempo, incluso en pacientes que lograron una respuesta completa al tratamiento. A veces se producen recaídas muchos años después del tratamiento. Por el momento, no hay ensayos aleatorizados que orienten a los médicos sobre la elección inicial de observación cautelosa, o el uso de rituximab, análogos de nucleósidos, alquilantes, quimioterapia combinada, anticuerpos monoclonales radiomarcados o combinaciones de estas opciones.; [Nivel de evidencia B1]
Para los pacientes con LNH de crecimiento lento no adyacente en estadios II y III, se ha propuesto la administración de radioterapia linfática central, pero no se recomienda su uso habitual como método de tratamiento.
Los pacientes con infección por el VHB resuelta (es decir, negativa para el antígeno de superficie del VHB, pero positiva para el anticuerpo central del VHB), presentan riesgo de reactivación de la infección por el VHB y necesitan vigilancia del DNA del VHB. La terapia profiláctica con nucleósidos redujo la reactivación de la infección por el VHB de un 10,8 % a un 2,1 % en un estudio retrospectivo de 326 pacientes.
Opciones de tratamiento del linfoma no Hodgkin de células B de crecimiento lento no adyacente en estadio II, III y IV
Las opciones de tratamiento del linfoma no Hodgkin de células B de crecimiento lento no adyacente en estadios II, III y IV son las siguientes:
- Observación cautelosa para pacientes asintomáticos.
- Rituximab solo o en combinación con citotóxicos utilizados en el tratamiento de primera línea.
- Lenalidomida y rituximab.
- Mantenimiento con rituximab.
- Obinutuzumab solo o en combinación con citotóxicos utilizados en el tratamiento de primera línea.
Debido a que ninguno de los tratamientos enumerados antes son curativos para la enfermedad en estadio avanzado, se encuentran en evaluación clínica varios abordajes innovadores.
Observación cautelosa para pacientes asintomáticos
La tasa de recaída es muy constante a lo largo del tiempo, incluso en pacientes que lograron una respuesta completa al tratamiento. De hecho, a veces se producen recaídas muchos años después del tratamiento. Para esta categoría, se puede considerar el tratamiento diferido (es decir, observación cautelosa hasta que el paciente presente síntomas antes de iniciar el tratamiento). El Follicular Lymphoma International Prognostic Index (FLIPI) y el FLIPI-2 revisado permiten predecir la supervivencia sin progresión (SSP) y la supervivencia general (SG), pero los puntajes no sirven para establecer la necesidad de tratamiento en pacientes asintomáticos.
Evidencia (observación cautelosa):
- En tres ensayos aleatorizados se comparó la observación cautelosa con la quimioterapia inmediata.; [Nivel de evidencia A1]
- En ninguno de los tres ensayos se observó una diferencia en la SG ni en la supervivencia por causa específica.
- En los pacientes asignados en forma aleatoria a observación cautelosa, la mediana de tiempo hasta la necesidad de tratamiento fue de 2 a 3 años; un tercio de los pacientes del grupo de observación cautelosa nunca necesitaron tratamiento (la mitad murió por otras causas y la otra mitad permaneció sin progresión después de 10 años).
- Un grupo seleccionado de 107 pacientes con linfoma folicular en estadio avanzado se trató con observación cautelosa inicial; el tratamiento posterior se retrasó durante una mediana de 55 meses. Estos pacientes lograron un tiempo sin fracaso del tratamiento y una SG equivalentes en comparación con una cohorte similar que recibió tratamiento inmediato con rituximab.[Nivel de evidencia C2] Esto implica que la observación cautelosa sigue siendo un abordaje relevante incluso en la era del rituximab.
Rituximab solo o en combinación con citotóxicos utilizados en el tratamiento de primera línea
El tratamiento estándar incluye rituximab, un anticuerpo monoclonal anti-CD20, solo como se usó en el ensayo ECOG-E4402 (NCT00075946), o en combinación con análogos de nucleósidos de purina, como fludarabina o cladribina, alquilantes (con o sin corticoesteroides) o quimioterapia combinada. El rituximab se puede considerar como tratamiento de primera línea, solo o en combinación con otros medicamentos. Es posible administrar el rituximab por vía intravenosa (IV) o subcutánea (SC); y en versiones biosimilares, como CT-P10 y GP2013, se ha observado eficacia e inocuidad equivalentes. Las combinaciones son las siguientes:
- R-bendamustina: rituximab y bendamustina.
- R-F: rituximab y fludarabina.
- R-CVP: rituximab, ciclofosfamida, vincristina y prednisona.
- R-CHOP: rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona. En un metanálisis de Cochrane, no se pudo identificar un beneficio para la SG al añadir doxorrubicina a los regímenes quimioterapéuticos con rituximab o sin este.[Nivel de evidencia A1]
- R-FM: rituximab, fludarabina y mitoxantrona.
- R-FCM: rituximab, fludarabina, ciclofosfamida y mitoxantrona.
Evidencia (rituximab con quimioterapia o sin esta):
- En un ensayo prospectivo aleatorizado con 534 pacientes de linfoma folicular en estadio avanzado sin tratamiento previo, se compararon los regímenes R-CHOP, R-FM y R-CVP.
- Después de una mediana de seguimiento de 84 meses, no hubo diferencia en la SG (tasa de SG a 8 años, 83 %, intervalo de confianza [IC] 95 %, 79–87 %), pero las tasas de SSP a 8 años favorecieron el tratamiento con R-CHOP (52 %) y R-FM (49 %) en comparación con R-CVP (42 %) (P para los 3 regímenes = 0,037).[Nivel de evidencia B1]
- En cuatro estudios prospectivos aleatorizados con pacientes sin tratamiento previo (que incluyeron a más de 1300 pacientes) y en un metanálisis de Cochrane que incluyó a pacientes con tratamiento previo y sin este (casi 1000 pacientes), el rituximab con quimioterapia combinada se comparó con la quimioterapia sola.; [Nivel de evidencia A1]
- El rituximab con quimioterapia fue superior en términos de supervivencia sin complicaciones (SSC) o SSP (osciló entre 2–3 años) en todos los estudios; esta combinación también fue superior en términos de SG en todos menos uno de los estudios (el beneficio absoluto osciló entre 6–13 % a los 4 años, P < 0,04; cociente de riesgos instantáneos [CRI], 0,63 [0,51–0,79] para el metanálisis).
- En todos los ensayos participaron pacientes sintomáticos que necesitaban tratamiento. Estos resultados no se oponen a la observación cautelosa cuando resulte apropiada.
- El resultado de la tomografía por emisión de positrones con flúor F 18-fludesoxiglucosa y tomografía computarizada (TEP-TC 18F-FDG) al finalizar el tratamiento con rituximab y terapia de inducción con quimioterapia es muy predictivo del desenlace. Aún no se sabe si decidir el tratamiento a partir del resultado de estas imágenes conduce a mejores desenlaces.
- En un ensayo aleatorizado prospectivo (NCT00991211), 527 pacientes con linfoma de crecimiento lento y linfoma de células de manto se asignaron de manera aleatoria para recibir bendamustina y rituximab o R-CHOP.[Nivel de evidencia B1]
- Después de una mediana de seguimiento de 45 meses, la mediana de SSP favoreció al grupo de bendamustina (69 vs. 31 meses [CRI, 0,58; IC 95 %, 0,44–0,74; P < 0,0001]), pero no hubo diferencia en la SG.
- El grupo de bendamustina se relacionó con tasas significativamente inferiores de alopecia, toxicidad hematológica, estomatitis, neuropatía periférica e infecciones que el grupo de R-CHOP.
- En un ensayo aleatorizado prospectivo similar, 447 pacientes con linfoma de células de manto de crecimiento lento se asignaron a bendamustina y rituximab versus R-CHOP o R-CVP.[Nivel de evidencia B1]
- Al cabo de una mediana de seguimiento de 65 meses, la tasa de SSP a 5 años favoreció al grupo de bendamustina y rituximab, 65,5 % versus 55,8 % (CRI, 0,61; IC 95 %, 0,45–0,85; P = 0,0025), pero no hubo diferencia en la SG.
- En el grupo de bendamustina y rituximab el aumento de las muertes por causas cardiovasculares (7 vs. 1) y por neoplasias malignas secundarias diferentes al linfoma (5 vs. 3) quizás contribuyó a la ausencia de ventaja en la SG.
Lenalidomida y rituximab
La combinación del inmunomodulador lenalidomida y rituximab (régimen R2) se propuso como un régimen alternativo a las combinaciones de citotóxicos, que acarrean efectos tóxicos subsecuentes a corto y largo plazo.
Evidencia (lenalidomida y rituximab):
- En un ensayo prospectivo aleatorizado (RELEVANCE [NCT01650701]) de 1030 pacientes con linfoma folicular sin tratamiento previo, se comparó el uso de rituximab y lenalidomida durante 18 meses con el uso de rituximab y quimioterapia (por lo general, R-CHOP). Todos los pacientes recibieron mantenimiento con rituximab hasta por 2 años.
- Al cabo de una mediana de seguimiento de 72 meses, la SSP a 6 años (60 y 59 %) y la SG a 3 años (89 %) fueron idénticas (CRI, 1,03; IC 95 %, 0,84–1,27; P = 0,78 para la SSP) (no se notificó el CRI para la SG).[Nivel de evidencia A1]
- En el ensayo se determinó que el régimen R2 es tan eficaz como el rituximab combinado con opciones de quimioterapia citotóxica. La tasa de transformación a linfoma de crecimiento rápido por año fue del 0,68 % en el grupo R2 y del 0,45 % en el grupo de quimioterapia R. Después de una mediana de seguimiento de 72 meses, no se observaron nuevas señales de seguridad.
- En un ensayo prospectivo aleatorizado de 358 pacientes con linfoma de crecimiento lento resistente al tratamiento (por lo general, linfoma folicular), el régimen R2 se comparó con el rituximab solo.
- Al cabo de una mediana de seguimiento de 28 meses, la mediana de SSP fue de 39,4 meses para el régimen R2 y de 14,1 meses para el rituximab solo (P < 0,0001), sin diferencias en la SG.[Nivel de evidencia B1]
Mantenimiento con rituximab
Luego de la terapia de inducción con rituximab solo o con rituximab y quimioterapia, es posible usar el rituximab 1 vez cada 2 o 3 meses como terapia de mantenimiento. Este abordaje se evaluó en varios estudios.
Evidencia (mantenimiento con rituximab en pacientes sin tratamiento previo):
- En el estudio PRIMA (NCT00140582), 1018 pacientes sintomáticos de riesgo alto sin tratamiento previo que alcanzaron una respuesta completa o una respuesta parcial después de la terapia de inducción con inmunoquimioterapia (por lo general, R-CHOP), se asignaron de manera aleatoria a 2 años de mantenimiento con rituximab o a no recibir mantenimiento.[Nivel de evidencia B1]
- Al cabo de una mediana de seguimiento de 9,0 años, la mediana de SSP favoreció al grupo de mantenimiento con rituximab, (10,5 años) en comparación con la observación (4,1 años) (CRI 0,61; IC 95 %, 0,52–0,73; P < 0,001), pero no hubo diferencia en la SG.
- En el estudio United Kingdom/International Study (NCT00112931), 379 pacientes asintomáticos con carga de enfermedad baja sin tratamiento previo, se asignaron de manera aleatoria a observación cautelosa versus inducción con rituximab, seguido de 2 años de mantenimiento con rituximab.[Nivel de evidencia A3]
- Si bien la SG y las tasas de transformación histológica no fueron diferentes a los 3 años, se favoreció el mantenimiento con rituximab con base a los resultados de los estudios por escalas de calidad de vida (CV) (Mental Adjustment to Cancer Scale, P = 0,0004 a los 7 meses, Illness Coping Score, P = 0,0012 a los 7 meses) y del tiempo transcurrido hasta el inicio de un tratamiento nuevo durante 3 años (54 % para la observación cautelosa vs. 12 % para el mantenimiento con rituximab [CRI, 0,21; IC 95 %, 0,14–0,31, P < 0,0001]).[Nivel de evidencia A3]
- En este estudio se indicó que para algunos pacientes la conducta expectante significó esperar preocupados. Sin embargo, desde la perspectiva de la SG y las tasas de transformación histológica, no se observó beneficio del mantenimiento con rituximab.
- En el estudio RESORT (NCT00075946), 289 pacientes asintomáticos con carga de enfermedad baja sin tratamiento previo se asignaron de manera aleatoria a inducción con rituximab solo, con una estrategia de reiterar el tratamiento con rituximab en el momento de la recaída, o inducción con rituximab y mantenimiento con rituximab cada 13 semanas hasta el fracaso terapéutico.[Nivel de evidencia A3]
- Después de una mediana de seguimiento de 4,5 años, no hubo diferencia en la mediana de tiempo hasta el fracaso terapéutico (definido como el fracaso con rituximab solo) ni en la CV relacionada con la salud. Cuando se usó una estrategia de reiterar el tratamiento con dosis significativamente más bajas de rituximab, se logró un control similar de la enfermedad.
En estos 3 ensayos aleatorizados de pacientes sin tratamiento previo, no se observó una ventaja del mantenimiento con rituximab en comparación con la observación y la terapia de reinducción en el momento de la recaída. En los ensayos se indica que hay un beneficio del mantenimiento con rituximab después de la reinducción para una recaída. Quedan muchas preguntas sin resolver sobre del mantenimiento con rituximab; en particular, aquellas relacionadas con la interrupción del tratamiento a los 2 años, así como su inocuidad y eficacia a largo plazo. En un ensayo en el que se prolongó el mantenimiento con rituximab a 5 años, se observaron una SSC y una SG similares en comparación con 1 año de mantenimiento tras la terapia de inducción con rituximab en pacientes sin tratamiento previo.[Nivel de evidencia A1]
- En el estudio FOLL12 ((NCT02063685) participaron 807 pacientes de linfoma folicular con carga tumoral alta sin tratamiento previo. Los pacientes recibieron rituximab con quimioterapia de inducción y se asignaron de manera aleatoria para recibir mantenimiento con rituximab estándar (cada 8 semanas por 2 años) o tratamiento posinducción (vigilancia, mantenimiento con rituximab o radioinmunoterapia) según la respuesta metabólica completa y el estado negativo para la enfermedad residual medible.
- Al cabo de una mediana de seguimiento de 53 meses, la tasa de SSP a 3 años fue del 86 % para los pacientes que recibieron el mantenimiento estándar y del 72 % para los pacientes que recibieron el tratamiento relacionado con la respuesta (P < 0,001). La SG a 3 años fue igual en ambos grupos (98 vs. 97 %; P = 0,238).[Nivel de evidencia B1]
- En este ensayo no se respalda el uso de una TEP-TC al final del tratamiento para orientar el mantenimiento con rituximab.
- En un ensayo prospectivo participaron 202 pacientes con linfoma folicular de baja carga tumoral que no se trató previamente. Los pacientes se asignaron al azar a recibir 4 dosis semanales de rituximab IV (dosis estándar, 375 mg/m2) o una dosis de rituximab IV seguida de 3 dosis semanales de rituximab SC (1400 mg) y dosis de mantenimiento en los meses 3, 5, 7 y 9.
- Al cabo de una mediana de seguimiento de 50,2 meses, la tasa de SSP a 4 años fue del 58,1 % (IC 95 %, 47,5–67,4%) en los pacientes del grupo SC que recibieron terapia de mantenimiento, y del 41,2 % (IC 95 %, 30,6–51,6 %) (CRI, 0,585; 0,939–0,871; P = 0,0076) en los pacientes del grupo IV que no recibieron terapia de mantenimiento.[Nivel de evidencia B1]
- No hubo diferencia en la SG ni en el tiempo transcurrido hasta el siguiente tratamiento.
En resumen, en todos los estudios de pacientes sin tratamiento previo, se observó una SSP mejor sin cambio en la SG.
Evidencia (mantenimiento con rituximab en pacientes con tratamiento previo):
- En un ensayo prospectivo aleatorizado de 465 pacientes con linfoma folicular en recaída, aquellos que respondieron al tratamiento con R-CHOP o CHOP se asignaron de manera aleatoria para recibir mantenimiento con rituximab (1 dosis cada 3 meses durante 2 años) o no recibir mantenimiento.[Nivel de evidencia B1]
- Al cabo de una mediana de seguimiento de 6 meses, el mantenimiento con rituximab mejoró la mediana de SSP (44 vs. 16 meses; P < 0,001), pero el beneficio para la SG a 5 años fue marginal (74 vs. 64 %, P = 0,07).
- El beneficio del mantenimiento fue evidente incluso para los pacientes que recibieron rituximab durante la terapia de inducción. La mayoría de los pacientes de ambos grupos recibieron más rituximab durante el tratamiento de rescate posterior al protocolo.
- En un ensayo prospectivo aleatorizado de 280 pacientes con linfoma folicular en recaída, aquellos que respondieron a la quimioterapia con consolidación de trasplante autógeno de células madre se asignaron de manera aleatoria para recibir 4 dosis de rituximab de mantenimiento o no recibir mantenimiento.[Nivel de evidencia B1]
- Al cabo de una mediana de seguimiento de 8,3 años, las tasas de SSP a 10 años favorecieron al grupo de mantenimiento (54 vs. 37 % [CRI 0,66; IC 95 %, 0,47–0,91, P = 0,012]), pero no hubo diferencia en la SG.
- En un metanálisis de 9 ensayos aleatorizados con 2586 pacientes de linfoma folicular, la mayoría en recaída, el mantenimiento con rituximab se comparó con la ausencia de mantenimiento, y en los pacientes con tratamiento previo se observó que el mantenimiento con rituximab mejoró la SG (CRIde muerte, 0,72; IC 95 %, 0,57-0,91).[Nivel de evidencia A1]
En pacientes con tratamiento previo hay más evidencia que indica una ventaja en la SG cuando se usa el mantenimiento con rituximab.
Obinutuzumab solo o en combinación con citotóxicos utilizados en el tratamiento de primera línea
El obinutuzumab es un anticuerpo monoclonal glucomodificado anti-CD20 de tipo II que produce más citotoxicidad celular dependiente de anticuerpos que el rituximab.
Evidencia (obinutuzumab):
- En un ensayo prospectivo aleatorizado (NCT01332968) con 1202 pacientes de linfoma folicular sin tratamiento previo, se comparó el uso de combinaciones de obinutuzumab o de rituximab con una de tres opciones de quimioterapia según la elección del investigador: el 50 % con bendamustina; el 33 % con ciclofosfamida, doxorrubicina, vincristina y prednisona (CHOP); el 10 % con ciclofosfamida, vincristina y prednisona (CVP). Después de 6 ciclos de terapia combinada, los pacientes recibieron 2 años de terapia de mantenimiento y recibieron el mismo anticuerpo cada 2 meses.
- Después de una mediana de seguimiento de 34,5 meses, la tasa de SSP a 3 años fue del 80 % en el grupo de obinutuzumab y del 73,3 % en el grupo de rituximab (CRI, 0,66; IC 95 %, 0,51–0,85, P = 0,001).[Nivel de evidencia B1]
- No hubo diferencia en la SG.
- La mortalidad por toxicidad fue más alta en los pacientes que usaron bendamustina en el grupo de obinutuzumab (5,6 %) y en el grupo de rituximab (4,4 %) en comparación con los resultados históricos. El número de muertes por toxicidad durante la terapia de primera línea parece excesivo en pacientes con linfoma de crecimiento lento de grado bajo y medianas de supervivencia que exceden los 15 años. En cambio, la mortalidad por toxicidad fue del 1 % al 2 % cuando cualquiera de los anticuerpos se combinó con CHOP o CVP.
Se han planteado varios problemas sobre este estudio:
- Los efectos secundarios (reacciones a la infusión y efectos adversos posteriores) fueron significativamente más altos con obinutuzumab.
- El obinutuzumab cuesta mucho más que el rituximab.
En resumen, en ausencia de modificaciones en la SG, resulta complicado decidir si se cambia el rituximab por obinutuzumab para combinarlo con quimioterapia en pacientes con linfoma folicular sin tratamiento previo. Las diferencias en la SSP quizás se pueden atribuir al desequilibrio en la dosificación del anticuerpo monoclonal; y el aumento de los efectos secundarios y del costo son factores mitigantes. En este ensayo, la bendamustina combinada con cualquier anticuerpo condujo a tasas de mortalidad tóxica inaceptables.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
References
- Hagenbeek A, Eghbali H, Monfardini S, et al.: Phase III intergroup study of fludarabine phosphate compared with cyclophosphamide, vincristine, and prednisone chemotherapy in newly diagnosed patients with stage III and IV low-grade malignant Non-Hodgkin's lymphoma. J Clin Oncol 24 (10): 1590-6, 2006.
- Gribben JG: How I treat indolent lymphoma. Blood 109 (11): 4617-26, 2007.
- Jacobs JP, Murray KJ, Schultz CJ, et al.: Central lymphatic irradiation for stage III nodular malignant lymphoma: long-term results. J Clin Oncol 11 (2): 233-8, 1993.
- Mendenhall NP, Million RR: Comprehensive lymphatic irradiation for stage II-III non-Hodgkin's lymphoma. Am J Clin Oncol 12 (3): 190-4, 1989.
- Kusumoto S, Arcaini L, Hong X, et al.: Risk of HBV reactivation in patients with B-cell lymphomas receiving obinutuzumab or rituximab immunochemotherapy. Blood 133 (2): 137-146, 2019.
- Eek R, Falkson G: The low-grade lymphoproliferative disorders. Oncology 54 (6): 441-58, 1997 Nov-Dec.
- Ardeshna KM, Smith P, Norton A, et al.: Long-term effect of a watch and wait policy versus immediate systemic treatment for asymptomatic advanced-stage non-Hodgkin lymphoma: a randomised controlled trial. Lancet 362 (9383): 516-22, 2003.
- Portlock CS, Rosenberg SA: No initial therapy for stage III and IV non-Hodgkin's lymphomas of favorable histologic types. Ann Intern Med 90 (1): 10-13, 1979.
- Solal-Céligny P, Roy P, Colombat P, et al.: Follicular lymphoma international prognostic index. Blood 104 (5): 1258-65, 2004.
- Federico M, Bellei M, Marcheselli L, et al.: Follicular lymphoma international prognostic index 2: a new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J Clin Oncol 27 (27): 4555-62, 2009.
- Brice P, Bastion Y, Lepage E, et al.: Comparison in low-tumor-burden follicular lymphomas between an initial no-treatment policy, prednimustine, or interferon alfa: a randomized study from the Groupe d'Etude des Lymphomes Folliculaires. Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 15 (3): 1110-7, 1997.
- Young RC, Longo DL, Glatstein E, et al.: The treatment of indolent lymphomas: watchful waiting v aggressive combined modality treatment. Semin Hematol 25 (2 Suppl 2): 11-6, 1988.
- Solal-Céligny P, Bellei M, Marcheselli L, et al.: Watchful waiting in low-tumor burden follicular lymphoma in the rituximab era: results of an F2-study database. J Clin Oncol 30 (31): 3848-53, 2012.
- Ghielmini M, Schmitz SF, Cogliatti SB, et al.: Prolonged treatment with rituximab in patients with follicular lymphoma significantly increases event-free survival and response duration compared with the standard weekly x 4 schedule. Blood 103 (12): 4416-23, 2004.
- Witzig TE, Vukov AM, Habermann TM, et al.: Rituximab therapy for patients with newly diagnosed, advanced-stage, follicular grade I non-Hodgkin's lymphoma: a phase II trial in the North Central Cancer Treatment Group. J Clin Oncol 23 (6): 1103-8, 2005.
- Hainsworth JD, Litchy S, Shaffer DW, et al.: Maximizing therapeutic benefit of rituximab: maintenance therapy versus re-treatment at progression in patients with indolent non-Hodgkin's lymphoma--a randomized phase II trial of the Minnie Pearl Cancer Research Network. J Clin Oncol 23 (6): 1088-95, 2005.
- Kahl BS, Hong F, Williams ME, et al.: Rituximab extended schedule or re-treatment trial for low-tumor burden follicular lymphoma: eastern cooperative oncology group protocol e4402. J Clin Oncol 32 (28): 3096-102, 2014.
- Buske C, Hiddemann W: Rituximab maintenance therapy in indolent NHL: a clinical review. Leuk Res 30 (Suppl 1): S11-5, 2006.
- Cartron G, Bachy E, Tilly H, et al.: Randomized Phase III Trial Evaluating Subcutaneous Rituximab for the First-Line Treatment of Low-Tumor Burden Follicular Lymphoma: Results of a LYSA Study. J Clin Oncol 41 (19): 3523-3533, 2023.
- Kim WS, Buske C, Ogura M, et al.: Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol 4 (8): e362-e373, 2017.
- Davies A, Merli F, Mihaljević B, et al.: Efficacy and safety of subcutaneous rituximab versus intravenous rituximab for first-line treatment of follicular lymphoma (SABRINA): a randomised, open-label, phase 3 trial. Lancet Haematol 4 (6): e272-e282, 2017.
- Jurczak W, Moreira I, Kanakasetty GB, et al.: Rituximab biosimilar and reference rituximab in patients with previously untreated advanced follicular lymphoma (ASSIST-FL): primary results from a confirmatory phase 3, double-blind, randomised, controlled study. Lancet Haematol 4 (8): e350-e361, 2017.
- Robinson KS, Williams ME, van der Jagt RH, et al.: Phase II multicenter study of bendamustine plus rituximab in patients with relapsed indolent B-cell and mantle cell non-Hodgkin's lymphoma. J Clin Oncol 26 (27): 4473-9, 2008.
- Rummel MJ, Niederle N, Maschmeyer G, et al.: Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 381 (9873): 1203-10, 2013.
- Flinn IW, van der Jagt R, Kahl B, et al.: First-Line Treatment of Patients With Indolent Non-Hodgkin Lymphoma or Mantle-Cell Lymphoma With Bendamustine Plus Rituximab Versus R-CHOP or R-CVP: Results of the BRIGHT 5-Year Follow-Up Study. J Clin Oncol 37 (12): 984-991, 2019.
- Czuczman MS, Koryzna A, Mohr A, et al.: Rituximab in combination with fludarabine chemotherapy in low-grade or follicular lymphoma. J Clin Oncol 23 (4): 694-704, 2005.
- Marcus R, Imrie K, Belch A, et al.: CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood 105 (4): 1417-23, 2005.
- Marcus R, Imrie K, Solal-Celigny P, et al.: Phase III study of R-CVP compared with cyclophosphamide, vincristine, and prednisone alone in patients with previously untreated advanced follicular lymphoma. J Clin Oncol 26 (28): 4579-86, 2008.
- Federico M, Luminari S, Dondi A, et al.: R-CVP versus R-CHOP versus R-FM for the initial treatment of patients with advanced-stage follicular lymphoma: results of the FOLL05 trial conducted by the Fondazione Italiana Linfomi. J Clin Oncol 31 (12): 1506-13, 2013.
- Luminari S, Ferrari A, Manni M, et al.: Long-Term Results of the FOLL05 Trial Comparing R-CVP Versus R-CHOP Versus R-FM for the Initial Treatment of Patients With Advanced-Stage Symptomatic Follicular Lymphoma. J Clin Oncol 36 (7): 689-696, 2018.
- Czuczman MS, Weaver R, Alkuzweny B, et al.: Prolonged clinical and molecular remission in patients with low-grade or follicular non-Hodgkin's lymphoma treated with rituximab plus CHOP chemotherapy: 9-year follow-up. J Clin Oncol 22 (23): 4711-6, 2004.
- Hainsworth JD, Litchy S, Morrissey LH, et al.: Rituximab plus short-duration chemotherapy as first-line treatment for follicular non-Hodgkin's lymphoma: a phase II trial of the Minnie Pearl Cancer Research Network. J Clin Oncol 23 (7): 1500-6, 2005.
- Hiddemann W, Kneba M, Dreyling M, et al.: Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 106 (12): 3725-32, 2005.
- Itchaki G, Gafter-Gvili A, Lahav M, et al.: Anthracycline-containing regimens for treatment of follicular lymphoma in adults. Cochrane Database Syst Rev 7: CD008909, 2013.
- Zinzani PL, Pulsoni A, Perrotti A, et al.: Fludarabine plus mitoxantrone with and without rituximab versus CHOP with and without rituximab as front-line treatment for patients with follicular lymphoma. J Clin Oncol 22 (13): 2654-61, 2004.
- Forstpointner R, Dreyling M, Repp R, et al.: The addition of rituximab to a combination of fludarabine, cyclophosphamide, mitoxantrone (FCM) significantly increases the response rate and prolongs survival as compared with FCM alone in patients with relapsed and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 104 (10): 3064-71, 2004.
- Herold M, Haas A, Srock S, et al.: Rituximab added to first-line mitoxantrone, chlorambucil, and prednisolone chemotherapy followed by interferon maintenance prolongs survival in patients with advanced follicular lymphoma: an East German Study Group Hematology and Oncology Study. J Clin Oncol 25 (15): 1986-92, 2007.
- Salles GA, Mounier N, de Guibert S, et al.: Rituximab combined with chemotherapy and interferon in follicular lymphoma patients: final analysis of the GELA-GOELAMS FL2000 study with a 5-year follow-up. [Abstract] Blood 110 (11): A-792, 2007.
- Schulz H, Bohlius J, Skoetz N, et al.: Combined immunochemotherapy with rituximab improves overall survival in patients with follicular and mantle cell lymphoma: updated meta-analysis results. [Abstract] Blood 108 (11): A-2760, 2006.
- Dupuis J, Berriolo-Riedinger A, Julian A, et al.: Impact of [(18)F]fluorodeoxyglucose positron emission tomography response evaluation in patients with high-tumor burden follicular lymphoma treated with immunochemotherapy: a prospective study from the Groupe d'Etudes des Lymphomes de l'Adulte and GOELAMS. J Clin Oncol 30 (35): 4317-22, 2012.
- Trotman J, Fournier M, Lamy T, et al.: Positron emission tomography-computed tomography (PET-CT) after induction therapy is highly predictive of patient outcome in follicular lymphoma: analysis of PET-CT in a subset of PRIMA trial participants. J Clin Oncol 29 (23): 3194-200, 2011.
- Morschhauser F, Fowler NH, Feugier P, et al.: Rituximab plus Lenalidomide in Advanced Untreated Follicular Lymphoma. N Engl J Med 379 (10): 934-947, 2018.
- Morschhauser F, Nastoupil L, Feugier P, et al.: Six-Year Results From RELEVANCE: Lenalidomide Plus Rituximab (R2) Versus Rituximab-Chemotherapy Followed by Rituximab Maintenance in Untreated Advanced Follicular Lymphoma. J Clin Oncol 40 (28): 3239-3245, 2022.
- Leonard JP, Trneny M, Izutsu K, et al.: AUGMENT: A Phase III Study of Lenalidomide Plus Rituximab Versus Placebo Plus Rituximab in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol 37 (14): 1188-1199, 2019.
- Bachy E, Seymour JF, Feugier P, et al.: Sustained Progression-Free Survival Benefit of Rituximab Maintenance in Patients With Follicular Lymphoma: Long-Term Results of the PRIMA Study. J Clin Oncol 37 (31): 2815-2824, 2019.
- Ardeshna KM, Qian W, Smith P, et al.: Rituximab versus a watch-and-wait approach in patients with advanced-stage, asymptomatic, non-bulky follicular lymphoma: an open-label randomised phase 3 trial. Lancet Oncol 15 (4): 424-35, 2014.
- Ansell SM: Follicular lymphoma: watch and wait is watch and worry. Lancet Oncol 15 (4): 368-9, 2014.
- Taverna C, Martinelli G, Hitz F, et al.: Rituximab Maintenance for a Maximum of 5 Years After Single-Agent Rituximab Induction in Follicular Lymphoma: Results of the Randomized Controlled Phase III Trial SAKK 35/03. J Clin Oncol 34 (5): 495-500, 2016.
- Luminari S, Manni M, Galimberti S, et al.: Response-Adapted Postinduction Strategy in Patients With Advanced-Stage Follicular Lymphoma: The FOLL12 Study. J Clin Oncol 40 (7): 729-739, 2022.
- van Oers MH, Tönnissen E, Van Glabbeke M, et al.: BCL-2/IgH polymerase chain reaction status at the end of induction treatment is not predictive for progression-free survival in relapsed/resistant follicular lymphoma: results of a prospective randomized EORTC 20981 phase III intergroup study. J Clin Oncol 28 (13): 2246-52, 2010.
- Pettengell R, Schmitz N, Gisselbrecht C, et al.: Rituximab purging and/or maintenance in patients undergoing autologous transplantation for relapsed follicular lymphoma: a prospective randomized trial from the lymphoma working party of the European group for blood and marrow transplantation. J Clin Oncol 31 (13): 1624-30, 2013.
- Vidal L, Gafter-Gvili A, Salles G, et al.: Rituximab maintenance for the treatment of patients with follicular lymphoma: an updated systematic review and meta-analysis of randomized trials. J Natl Cancer Inst 103 (23): 1799-806, 2011.
- Marcus R, Davies A, Ando K, et al.: Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N Engl J Med 377 (14): 1331-1344, 2017.
Tratamiento del linfoma no Hodgkin de células B de crecimiento lento recidivante
En general, el tratamiento con fármacos estándar rara vez produce la curación de pacientes con linfoma no Hodgkin (LNH) de células B en recaída. A menudo se obtienen remisiones prolongadas después de una recaída en pacientes con linfomas de crecimiento lento, pero por lo general vuelven a recaer. Una supervivencia favorable después de la recaída se relacionó con una edad menor de 60 años, remisión completa en lugar de remisión parcial y una respuesta con más de 2 años de duración. Sin embargo, incluso los pacientes del subconjunto más favorable tienen una mortalidad diez veces mayor en comparación con las tasas de la población de los Estados Unidos ajustadas por edad.
Los pacientes con linfoma de crecimiento lento que recaen, a menudo logran controlar su enfermedad con quimioterapia en monoterapia o quimioterapia combinada, rituximab (un anticuerpo monoclonal anti-CD20), lenalidomida, anticuerpos monoclonales anti-CD20 radiomarcados o radioterapia paliativa. No obstante, la supervivencia a largo plazo sin una segunda recaída es poco común y por lo general se presentan múltiples recaídas. Los pacientes con linfoma de crecimiento lento a veces recaen con un tipo histológico más maligno. Si el modelo clínico de recaída indica que la enfermedad tiene un comportamiento más maligno, se obtiene una biopsia. Si se confirma que la enfermedad se convirtió a un tipo histológico más agresivo, se debe cambiar el tratamiento a un régimen que corresponda al tipo histológico. El crecimiento rápido o discordante en diversos sitios de la enfermedad quizás indique una conversión histológica.
En una revisión retrospectiva de 325 pacientes atendidos entre 1972 y 1999, el riesgo de transformación histológica fue del 30 % a los 10 años del diagnóstico. En esta serie, los factores de riesgo alto de transformación histológica posterior fueron estadio avanzado, riesgo alto en el Follicular Lymphoma International Prognostic Index y conducta expectante. La mediana de supervivencia después de la transformación fue de 1 a 2 años, con un 25 % de los pacientes vivos a los 5 años y cerca del 10 % al 20 % de los pacientes vivos 10 años después del segundo tratamiento.
En un ensayo prospectivo con 631 pacientes de linfoma folicular, al cabo de una mediana de seguimiento de 60 meses en la era de rituximab (2002–2009), se encontró que la tasa de transformación a un tipo histológico de grado más alto a 5 años fue del 11 %. La mediana de supervivencia general (SG) después de la transformación fue de 50 meses y la tasa de SG a 5 años fue del 66 % cuando la transformación se produjo más de 18 meses después del diagnóstico de linfoma folicular. En esta serie, se describió un pronóstico mejor para los pacientes con transformación que para los pacientes tratados antes de la era del rituximab.
Para obtener información sobre descripciones de los regímenes que se usan para tratar las conversiones histológicas, consultar la sección Tratamiento del linfoma no Hodgkin de células B de crecimiento rápido recidivante. La duración de la segunda remisión a veces es breve y es posible considerar la participación en ensayos clínicos.
Opciones de tratamiento del linfoma no Hodgkin de células B de crecimiento lento recidivante
Las opciones de tratamiento del linfoma no Hodgkin de células B de crecimiento lento recidivante son las siguientes:
- Rituximab solo o en combinación con citotóxicos utilizados en el tratamiento de primera línea.
- Obinutuzumab solo o en combinación con citotóxicos utilizados en el tratamiento de primera línea.
- Lenalidomida y rituximab.
- Inhibidor de EZH2.
- Tazemetostat.
- Captadores biespecíficos de células T.
- Mosunetuzumab.
- Terapia de células T con receptor de antígeno quimérico.
- Trasplante de células madre.
- Inhibidor de la fosfatidilinositol-3–cinasa.
- Copanlisib.
- Radioterapia paliativa.
Rituximab solo o en combinación con citotóxicos utilizados en el tratamiento de primera línea
El rituximab produce una tasa de respuesta del 40 % al 50 % en los pacientes que recaen con linfomas de células B de crecimiento lento. El rituximab también se puede administrar con quimioterapia combinada.
Evidencia (rituximab):
- En 3 estudios prospectivos aleatorizados de pacientes con linfoma de crecimiento lento recidivante y con tratamiento previo, los pacientes se asignaron de manera aleatoria para recibir mantenimiento con rituximab después de un segundo tratamiento con quimioterapia combinada (con rituximab o sin este durante la inducción) o rituximab solo.
- En todos los ensayos se observó una prolongación de la duración de la respuesta. En un ensayo con una mediana de seguimiento de 39 meses, se observó una mejora en la mediana de supervivencia sin progresión (SSP) (3,7 vs. 1,3 años, P < 0,001) y la tasa de SG a 5 años (74 vs. 64 %, P = 0,07), que favoreció el mantenimiento con rituximab.
Obinutuzumab solo o en combinación con citotóxicos utilizados en el tratamiento de primera línea
El obinutuzumab es un anticuerpo monoclonal que se une a CD20 y tiene un epítopo alternativo de unión.
Evidencia (obinutuzumab):
- En un ensayo prospectivo aleatorizado (NCT01059630) de 396 pacientes con linfoma de crecimiento lento resistente al tratamiento con rituximab (en su mayoría linfoma folicular), los pacientes recibieron obinutuzumab con bendamustina seguido de terapia de mantenimiento con obinutuzumab durante 2 años versus bendamustina sola sin terapia de mantenimiento.[Nivel de evidencia A1]
- Después de una mediana de seguimiento de 31,8 meses, la combinación de obinutuzumab favoreció la tasa de SG a 2 años (74,5 vs. 65,1 %) (cociente de riesgos instantáneos [CRI], 0,67; intervalo de confianza [IC] 95 %, 0,47–0,96;P < 0,027). La mediana de SSP también favoreció la combinación de obinutuzumab (25,8 meses [IC 95 %, 19,5–41,1] vs. 14,1 meses [IC 95 %, 12,6–16,0]) (CRI, 0,57; IC 95 %, 0,44–0,73; P < 0,001).[Nivel de evidencia A1]
- Este diseño no permitió evaluar la contribución de la terapia de mantenimiento al desenlace.
Lenalidomida y rituximab
Se notificaron respuestas a la lenalidomida en el 20 % al 56 % de los pacientes; en particular en aquellos con linfoma folicular y linfoma linfocítico de células pequeñas; se observaron respuestas aún mayores para la combinación de lenalidomida y rituximab.[Nivel de evidencia C3]
Inhibidor de EZH2
Tazemetostat
El tazemetostat es un inhibidor de EZH2, una histona–metiltransferasa necesaria para la formación de los centros germinativos de los ganglios linfáticos, en especial, cuando hay mutaciones activadoras en EZH2.
Evidencia (tazemetostat):
- En un estudio de fase II, participaron 99 pacientes con linfoma folicular en recaída o resistente al tratamiento, 45 presentaron mutaciones activadoras en EZH2 y 54 presentaron EZH2 de tipo natural (normal).
- El tratamiento con tazemetostat produjo una tasa de respuesta objetiva del 69 % (IC 95 %, 53–82 %) en los pacientes con mutaciones activadoras versus un 35 % (IC 95 %, 23–49%) en los pacientes con EZH2 de tipo natural.
- Al cabo de una mediana de seguimiento de 22 meses, la mediana de SSP fue de 13,8 meses (IC 95 %, 10,7–22,0) en los pacientes con mutaciones activadoras y de 11,1 meses (IC 95 %, 3,7–14,6) en los pacientes con EZH2 de tipo natural.[Nivel de evidencia C3]
- Se observaron efectos adversos relacionados con el tratamiento de grado 3 o 4 en el 4 % de los pacientes.
Captadores biespecíficos de células T
Los captadores biespecíficos de células T se unen a CD20 (o CD19) y a CD3 para dar instrucciones a las células T de eliminar o destruir los linfocitos B malignos. De manera similar a la terapia de células T con CAR, casi la mitad de los pacientes que reciben esta terapia experimentan el síndrome de liberación de citocinas.
Mosunetuzumab
Evidencia (mosunetuzumab):
- En un estudio de fase II multicéntrico y de un solo grupo, 90 pacientes con linfoma folicular en recaída o resistente al tratamiento después de 2 o más líneas de tratamiento anteriores (incluso una terapia anti-CD20 y un alquilante) recibieron mosunetuzumab.
- Al cabo de una mediana de seguimiento de 27 meses, la tasa de respuesta objetiva fue del 77,8 % (IC 95 %, 67,8–85,9 %) y la tasa de respuesta completa fue del 60,0 % (IC 95 %, 49,1–70,2 %), según la evaluación del investigador. No se alcanzó la mediana de SSP (según la evaluación del investigador). La SSP a 24 meses fue de 51,4 meses (IC 95 %, 39,4–63,3) después de recibir mosunetuzumab, en comparación con 23,5 meses (IC 95 %, 14,5–32,5) para el último tratamiento previo de los pacientes (quimioinmunoterapia 63 %, inhibidor de PI3K 8 %, terapia de células T con CAR 2 %, anticuerpo anti-CD20 con lenalidomida y otros 2 %).
- El síndrome de liberación de citocinas se presentó en el 44,4 % de los pacientes; en el 97,2 % de los casos fue de grado 1 o 2.[Nivel de evidencia C3]
Terapia de células T con receptor de antígeno quimérico
La terapia de células T con receptor de antígeno quimérico (CAR), con la terapia autógena anti-CD19 de axicabtagén ciloleucel, lisocabtagén maraleucel o tisagenlecleucel, se aprobó para los pacientes con linfoma folicular recidivante después de recibir 2 o más líneas de tratamiento previo.
Evidencia (terapia de células T con CAR):
- En un ensayo de fase II, 153 pacientes con linfoma folicular o linfoma de zona marginal en recaída o resistente al tratamiento recibieron axicabtagén ciloleucel.
- Después de una mediana de seguimiento de 17,5 meses, la tasa de respuesta general fue del 92 % (IC 95 %, 85–97 %) y la tasa de respuesta completa fue del 74 %.
- La tasa de SSP a 18 meses fue del 64,8 % (IC 95 %, 54,2–73,5 %).[Nivel de evidencia C3]
- El síndrome de liberación de citocinas se presentó en el 78 % de los pacientes y fue de grado 3 o 4 en el 6 % de los pacientes.
- De todos los pacientes, el 50 % necesitó tocilizumab y el 5 % vasopresores. Se presentaron episodios neurológicos de grado 3 o 4 en el 15 % de los pacientes.
- En un ensayo de fase II, 98 pacientes con linfoma folicular en recaída o resistente al tratamiento después de 2 o más líneas de tratamiento previo recibieron terapia de células T con CAR anti-CD19 con tisagenlecleucel.
- Al cabo de una mediana de seguimiento de 16,6 meses, la tasa de respuesta completa fue del 69,1 % (IC 95 %, 58,8–78,3 %) y la tasa de respuesta general fue del 86,2 % (77,5–92,4 %).[Nivel de evidencia C3]
- El síndrome de liberación de citocinas de grado 3 o 4 se presentó en el 48,5 % de los pacientes y el 37,1 % presentó neurotoxicidad de grado 3 o 4.
Las células T con CAR se usan en pacientes de riesgo alto que presentaron recaída de la enfermedad de forma rápida tras la quimioinmunoterapia. Este abordaje se considera en el contexto de muchos otros fármacos disponibles.
Trasplante de células madre
En muchas instituciones, se utilizan TCM autógenos o alogénicos para pacientes de riesgo alto con recaída rápida de la enfermedad tras la quimioinmunoterapia. Este abordaje se considera en el contexto de otros numerosos fármacos disponibles.
Evidencia (trasplante de células madre):
- En el German Low-Grade Lymphoma Study Group, 307 pacientes con linfoma folicular recibieron 2 ciclos de quimioterapia de inducción similar al régimen CHOP (ciclofosfamida, doxorrubicina, vincristina y prednisona) y, a continuación, se asignaron de manera aleatoria para recibir un trasplante de células madre (TCM) autógeno versus mantenimiento con interferón.
- Al cabo de una mediana de seguimiento de 4,2 años, la tasa de SSP a 5 años fue del 65 % en los pacientes que recibieron trasplante versus el 33 % en los pacientes que recibieron interferón (P < 0,001). No hubo diferencia en la SG.[Nivel de evidencia B1]
Inhibidor de la fosfatidilinositol-3–cinasa (PI3K)
Copanlisib
Evidencia (copanlisib):
- En un estudio aleatorizado, con enmascaramiento doble y controlado con placebo, se asignaron 307 pacientes con linfoma de crecimiento lento en recaída a recibir copanlisib con rituximab y 151 pacientes a recibir placebo con rituximab (aleatorización de 2:1).
- Al cabo de una mediana de seguimiento de 19,2 meses, la mediana de SSP fue de 21,5 meses en los pacientes del grupo de copanlisib con rituximab (IC 95 %, 17,8–33,0) y de 13,8 meses (IC 95 %, 10,2–17,5) en los pacientes del grupo de placebo con rituximab (CRI, 0,52; IC 95 %, 0,39–0,69; P < 0,0001).[Nivel de evidencia B1]
- Los efectos adversos de grado 3 a 4 más comunes fueron la hiperglucemia y la hipertensión, los cuales se presentaron en la mitad de los pacientes que recibieron copanlisib.
- En un estudio de fase II se incluyeron 142 pacientes con linfoma de crecimiento lento en recaída o resistente al tratamiento.
- El tratamiento con copanlisib intravenoso produjo una tasa de respuesta objetiva del 59 % (respuesta completa del 12 %) y una mediana de duración de la respuesta de 22,6 meses.[Nivel de evidencia C3]
Los inhibidores de PI3K tienen efectos adversos importantes que incluyen neumonitis, colitis, transaminitis, hipertensión, hiperglucemia, exantema y aumento del riesgo de infecciones. Los efectos adversos han afectado el uso de estos fármacos como tratamiento inicial hasta que en ensayos confirmatorios se pueda establecer su eficacia en el uso de combinaciones terapéuticas. En la actualidad, estos fármacos están aprobados para el tratamiento del linfoma folicular en recaída o resistente al tratamiento después de 2 líneas de terapia previas.
Radioterapia paliativa
Se puede lograr paliación con dosis muy bajas (4 Gy) de radioterapia dirigida al campo comprometido en 2 fracciones en pacientes con recaídas de la enfermedad de crecimiento rápido y lento. En un ensayo aleatorizado prospectivo, el tratamiento con 4 Gy fue inferior al tratamiento con 24 Gy en 12 fracciones para la SSP (77 vs. 92 %, P < 0,0001).[Nivel de evidencia B1]
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
References
- Casulo C, Byrtek M, Dawson KL, et al.: Early Relapse of Follicular Lymphoma After Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone Defines Patients at High Risk for Death: An Analysis From the National LymphoCare Study. J Clin Oncol 33 (23): 2516-22, 2015.
- Weisdorf DJ, Andersen JW, Glick JH, et al.: Survival after relapse of low-grade non-Hodgkin's lymphoma: implications for marrow transplantation. J Clin Oncol 10 (6): 942-7, 1992.
- Peterson BA: Current treatment of follicular low-grade lymphomas. Semin Oncol 26 (5 Suppl 14): 2-11, 1999.
- Haas RL, Poortmans P, de Jong D, et al.: High response rates and lasting remissions after low-dose involved field radiotherapy in indolent lymphomas. J Clin Oncol 21 (13): 2474-80, 2003.
- Tsimberidou AM, O'Brien S, Khouri I, et al.: Clinical outcomes and prognostic factors in patients with Richter's syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J Clin Oncol 24 (15): 2343-51, 2006.
- Montoto S, Davies AJ, Matthews J, et al.: Risk and clinical implications of transformation of follicular lymphoma to diffuse large B-cell lymphoma. J Clin Oncol 25 (17): 2426-33, 2007.
- Yuen AR, Kamel OW, Halpern J, et al.: Long-term survival after histologic transformation of low-grade follicular lymphoma. J Clin Oncol 13 (7): 1726-33, 1995.
- Link BK, Maurer MJ, Nowakowski GS, et al.: Rates and outcomes of follicular lymphoma transformation in the immunochemotherapy era: a report from the University of Iowa/MayoClinic Specialized Program of Research Excellence Molecular Epidemiology Resource. J Clin Oncol 31 (26): 3272-8, 2013.
- Davis TA, White CA, Grillo-López AJ, et al.: Single-agent monoclonal antibody efficacy in bulky non-Hodgkin's lymphoma: results of a phase II trial of rituximab. J Clin Oncol 17 (6): 1851-7, 1999.
- Piro LD, White CA, Grillo-López AJ, et al.: Extended Rituximab (anti-CD20 monoclonal antibody) therapy for relapsed or refractory low-grade or follicular non-Hodgkin's lymphoma. Ann Oncol 10 (6): 655-61, 1999.
- Davis TA, Grillo-López AJ, White CA, et al.: Rituximab anti-CD20 monoclonal antibody therapy in non-Hodgkin's lymphoma: safety and efficacy of re-treatment. J Clin Oncol 18 (17): 3135-43, 2000.
- Hainsworth JD, Litchy S, Shaffer DW, et al.: Maximizing therapeutic benefit of rituximab: maintenance therapy versus re-treatment at progression in patients with indolent non-Hodgkin's lymphoma--a randomized phase II trial of the Minnie Pearl Cancer Research Network. J Clin Oncol 23 (6): 1088-95, 2005.
- Lockmer S, Østenstad B, Hagberg H, et al.: Chemotherapy-Free Initial Treatment of Advanced Indolent Lymphoma Has Durable Effect With Low Toxicity: Results From Two Nordic Lymphoma Group Trials With More Than 10 Years of Follow-Up. J Clin Oncol : JCO1800262, 2018.
- Forstpointner R, Dreyling M, Repp R, et al.: The addition of rituximab to a combination of fludarabine, cyclophosphamide, mitoxantrone (FCM) significantly increases the response rate and prolongs survival as compared with FCM alone in patients with relapsed and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 104 (10): 3064-71, 2004.
- Canellos GP: CHOP may have been part of the beginning but certainly not the end: issues in risk-related therapy of large-cell lymphoma. J Clin Oncol 15 (5): 1713-6, 1997.
- van Oers MH, Van Glabbeke M, Giurgea L, et al.: Rituximab maintenance treatment of relapsed/resistant follicular non-Hodgkin's lymphoma: long-term outcome of the EORTC 20981 phase III randomized intergroup study. J Clin Oncol 28 (17): 2853-8, 2010.
- van Oers MH, Klasa R, Marcus RE, et al.: Rituximab maintenance improves clinical outcome of relapsed/resistant follicular non-Hodgkin lymphoma in patients both with and without rituximab during induction: results of a prospective randomized phase 3 intergroup trial. Blood 108 (10): 3295-301, 2006.
- Martinelli G, Schmitz SF, Utiger U, et al.: Long-term follow-up of patients with follicular lymphoma receiving single-agent rituximab at two different schedules in trial SAKK 35/98. J Clin Oncol 28 (29): 4480-4, 2010.
- Sehn LH, Chua N, Mayer J, et al.: Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol 17 (8): 1081-93, 2016.
- Cheson BD, Chua N, Mayer J, et al.: Overall Survival Benefit in Patients With Rituximab-Refractory Indolent Non-Hodgkin Lymphoma Who Received Obinutuzumab Plus Bendamustine Induction and Obinutuzumab Maintenance in the GADOLIN Study. J Clin Oncol 36 (22): 2259-2266, 2018.
- Witzig TE, Wiernik PH, Moore T, et al.: Lenalidomide oral monotherapy produces durable responses in relapsed or refractory indolent non-Hodgkin's Lymphoma. J Clin Oncol 27 (32): 5404-9, 2009.
- Leonard JP, Jung SH, Johnson J, et al.: Randomized Trial of Lenalidomide Alone Versus Lenalidomide Plus Rituximab in Patients With Recurrent Follicular Lymphoma: CALGB 50401 (Alliance). J Clin Oncol 33 (31): 3635-40, 2015.
- Morschhauser F, Tilly H, Chaidos A, et al.: Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol 21 (11): 1433-1442, 2020.
- Budde LE, Sehn LH, Matasar M, et al.: Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single-arm, multicentre, phase 2 study. Lancet Oncol 23 (8): 1055-1065, 2022.
- Hutchings M, Mous R, Clausen MR, et al.: Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an open-label, phase 1/2 study. Lancet 398 (10306): 1157-1169, 2021.
- Bartlett NL, Sehn LH, Matasar MJ, et al.: Mosunetuzumab monotherapy demonstrates durable efficacy with a manageable safety profile in patients with relapsed/refractory follicular lymphoma who received ≥2 prior therapies: updated results from a pivotal phase II study. [Abstract] Blood 140 (Suppl 1): A-610, 1467-70, 2022.
- Jacobson CA, Chavez JC, Sehgal AR, et al.: Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol 23 (1): 91-103, 2022.
- Fowler NH, Dickinson M, Dreyling M, et al.: Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med 28 (2): 325-332, 2022.
- Freedman A, Friedberg JW, Gribben J: High-dose therapy for follicular lymphoma. Oncology (Huntingt) 14 (3): 321-6, 329; discussion 330-2, 338, 2000.
- Brice P, Simon D, Bouabdallah R, et al.: High-dose therapy with autologous stem-cell transplantation (ASCT) after first progression prolonged survival of follicular lymphoma patients included in the prospective GELF 86 protocol. Ann Oncol 11 (12): 1585-90, 2000.
- Khouri IF, McLaughlin P, Saliba RM, et al.: Eight-year experience with allogeneic stem cell transplantation for relapsed follicular lymphoma after nonmyeloablative conditioning with fludarabine, cyclophosphamide, and rituximab. Blood 111 (12): 5530-6, 2008.
- Sebban C, Brice P, Delarue R, et al.: Impact of rituximab and/or high-dose therapy with autotransplant at time of relapse in patients with follicular lymphoma: a GELA study. J Clin Oncol 26 (21): 3614-20, 2008.
- Thomson KJ, Morris EC, Milligan D, et al.: T-cell-depleted reduced-intensity transplantation followed by donor leukocyte infusions to promote graft-versus-lymphoma activity results in excellent long-term survival in patients with multiply relapsed follicular lymphoma. J Clin Oncol 28 (23): 3695-700, 2010.
- Lenz G, Dreyling M, Schiegnitz E, et al.: Myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission prolongs progression-free survival in follicular lymphoma: results of a prospective, randomized trial of the German Low-Grade Lymphoma Study Group. Blood 104 (9): 2667-74, 2004.
- Matasar MJ, Capra M, Özcan M, et al.: Copanlisib plus rituximab versus placebo plus rituximab in patients with relapsed indolent non-Hodgkin lymphoma (CHRONOS-3): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 22 (5): 678-689, 2021.
- Dreyling M, Santoro A, Mollica L, et al.: Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol 35 (35): 3898-3905, 2017.
- Haas RL, Poortmans P, de Jong D, et al.: Effective palliation by low dose local radiotherapy for recurrent and/or chemotherapy refractory non-follicular lymphoma patients. Eur J Cancer 41 (12): 1724-30, 2005.
- Hoskin PJ, Kirkwood AA, Popova B, et al.: 4 Gy versus 24 Gy radiotherapy for patients with indolent lymphoma (FORT): a randomised phase 3 non-inferiority trial. Lancet Oncol 15 (4): 457-63, 2014.
Tratamiento del linfoma no Hodgkin de células B de crecimiento rápido en estadio I y del linfoma no Hodgkin de células B de crecimiento rápido adyacente en estadio II
Los pacientes con linfoma difuso de células B grandes (LDCBG) de crecimiento rápido en estadio I o LDCBG de crecimiento rápido adyacente en estadio II son aptos para recibir quimioterapia combinada con radioterapia dirigida al campo comprometido (RTCC) o sin esta.
Los pacientes con infección por el virus de la hepatitis B (VHB) resuelta (es decir, negativa para el antígeno de superficie del VHB, pero positiva para el anticuerpo central del VHB), presentan riesgo de reactivación de la infección por el VHB y necesitan vigilancia del DNA del VHB. La terapia profiláctica con nucleósidos redujo la reactivación de la infección por el VHB de un 10,8 % a un 2,1 % en un estudio retrospectivo de 326 pacientes.
Opciones de tratamiento del linfoma no Hodgkin de células B de crecimiento rápido en estadio I y el linfoma no Hodgkin de células B de crecimiento rápido adyacente en estadio II
Las opciones de tratamiento del linfoma no Hodgkin de células B de crecimiento rápido en estadio I y el linfoma no Hodgkin de células B de crecimiento rápido adyacente en estadio II son las siguientes:
- Rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona con radioterapia dirigida al campo comprometido o sin esta.
- Rituximab, doxorrubicina, ciclofosfamida, vindesina, bleomicina, prednisona (R-ACVBP) (en evaluación clínica).
Rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona con radioterapia dirigida al campo comprometido o sin esta
A partir de la confirmación de la eficacia del rituximab para la enfermedad en estadio avanzado se planteó el uso de rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona con radioterapia dirigida al campo comprometido o sin esta (R-CHOP) con radioterapia o sin esta, pero este uso solo se respalda en comparaciones retrospectivas.[Nivel de evidencia C2]
- R-CHOP (4 a 6 ciclos).
- R-CHOP (3 a 6 ciclos) y RTCC.
Evidencia (rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona con radioterapia dirigida al campo comprometido o sin esta):
- En un ensayo prospectivo aleatorizado de 334 pacientes con LDCBG sin gran masa tumoral (≤7 cm) en estadio I o estadio II, los pacientes recibieron 4 a 6 ciclos de R-CHOP-14 (R-CHOP administrado cada 2 semanas) y luego se asignaron de manera aleatoria para recibir 40 Gy de radioterapia o a no recibir radioterapia.
- Después de una mediana de seguimiento de 64 meses, la supervivencia sin complicaciones a 5 años (89–92 %, P = 0,18) y la supervivencia general (SG) a 5 años (92–96 %, P = 0,32) fueron las mismas.[Nivel de evidencia A1]
De manera semejante a los resultados de los estudios aleatorizados de radioterapia antes de la era de prerituximab, es posible diferir la radioterapia para los pacientes sin gran masa tumoral en estadios tempranos. Para los pacientes que no toleran un curso prolongado de quimioterapia, se obtienen resultados equivalentes con 3 ciclos de R-CHOP y radioterapia de acuerdo a ensayos retrospectivos de un solo grupo.
- Después de una mediana de seguimiento de 64 meses, la supervivencia sin complicaciones a 5 años (89–92 %, P = 0,18) y la supervivencia general (SG) a 5 años (92–96 %, P = 0,32) fueron las mismas.[Nivel de evidencia A1]
- En un ensayo prospectivo aleatorizado (NCT00278421) de 592 pacientes menores de 60 años con LDCBG sin gran masa tumoral (<7,5 cm) en estadio I o estadio II, los pacientes se asignaron de manera aleatoria para recibir 4 o 6 ciclos de R-CHOP (con 2 ciclos más de rituximab después de 4 ciclos).
- Después de una mediana de seguimiento de 66 meses, la tasa de supervivencia sin progresión (SSP) a 3 años fue del 96 % (IC 95 %, 94−99 %) para los pacientes que recibieron 4 ciclos de R-CHOP, esta fue un 3 % mejor (el límite inferior del valor unilateral de IC 95 % fue 0) que la tasa de SSP de los pacientes que recibieron 6 ciclos, lo que estableció una ausencia de inferioridad para el régimen de 4 ciclos.[Nivel de evidencia B1]
- En un análisis retrospectivo realizado en el Memorial Sloan Kettering Cancer Center entre 2001 y 2015, se incluyeron 341 pacientes con enfermedad en estadio I. En el análisis se observó que el 66 % de los pacientes tenían una presentación extraganglionar. Después de R-CHOP (o un régimen similar), con radioterapia o sin esta, la tasa de supervivencia sin enfermedad a 5 años fue del 77 %, y la tasa de SG a 5 años fue del 94 %.[Nivel de evidencia C3] En un análisis multivariante se indicó que la radioterapia puede mejorar los desenlaces de los pacientes con enfermedad extraganglionar y positiva para el compromiso en la tomografía por emisión de positrones (TEP) al final del tratamiento. Esta hipótesis debe confirmarse en un ensayo prospectivo aleatorizado.
En resumen, para los pacientes de LDCBG sin gran masa tumoral (<7 cm) en estadio I o estadio II y con pronóstico favorable, 4 ciclos de R-CHOP son suficientes. En los pacientes con pronóstico desfavorable, se pueden administrar 6 ciclos de R-CHOP o 3 ciclos de R-CHOP y 40 Gy de radioterapia. Los pacientes en estadio temprano con gran masa tumoral (>7,5 cm) no se han estudiado en ensayos aleatorizados; por lo general se administra una terapia de modalidad combinada con 4 a 6 ciclos de R-CHOP y radioterapia. Aunque en un estudio retrospectivo se indicó que los pacientes con enfermedad extraganglionar en estadio I y una TEP positiva al final del tratamiento se pueden beneficiar de la radioterapia, esta hipótesis se debe confirmar en un ensayo prospectivo aleatorizado.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
References
- Kusumoto S, Arcaini L, Hong X, et al.: Risk of HBV reactivation in patients with B-cell lymphomas receiving obinutuzumab or rituximab immunochemotherapy. Blood 133 (2): 137-146, 2019.
- Reyes F, Lepage E, Ganem G, et al.: ACVBP versus CHOP plus radiotherapy for localized aggressive lymphoma. N Engl J Med 352 (12): 1197-205, 2005.
- Ketterer N, Coiffier B, Thieblemont C, et al.: Phase III study of ACVBP versus ACVBP plus rituximab for patients with localized low-risk diffuse large B-cell lymphoma (LNH03-1B). Ann Oncol 24 (4): 1032-7, 2013.
- Persky DO, Unger JM, Spier CM, et al.: Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. J Clin Oncol 26 (14): 2258-63, 2008.
- Lamy T, Damaj G, Soubeyran P, et al.: R-CHOP 14 with or without radiotherapy in nonbulky limited-stage diffuse large B-cell lymphoma. Blood 131 (2): 174-181, 2018.
- Poeschel V, Held G, Ziepert M, et al.: Four versus six cycles of CHOP chemotherapy in combination with six applications of rituximab in patients with aggressive B-cell lymphoma with favourable prognosis (FLYER): a randomised, phase 3, non-inferiority trial. Lancet 394 (10216): 2271-2281, 2019.
- Bobillo S, Joffe E, Lavery JA, et al.: Clinical characteristics and outcomes of extranodal stage I diffuse large B-cell lymphoma in the rituximab era. Blood 137 (1): 39-48, 2021.
Tratamiento del linfoma no Hodgkin de células B de crecimiento rápido no adyacente en estadio II, III y IV
El tratamiento aceptado para los pacientes con linfoma no Hodgkin (LNH) de crecimiento rápido en estadios avanzados es la quimioterapia combinada, sola o que se complementa con radioterapia dirigida al campo local.
- Pola-R-CHP: polatuzumab vedotina + rituximab + ciclofosfamida + doxorrubicina + prednisona.
- R-CHOP: rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona.
- R-ACVBP: rituximab, un anticuerpo monoclonal anti-CD20, doxorrubicina, ciclofosfamida, vindesina, bleomicina y prednisona.
Opciones de tratamiento del linfoma no Hodgkin de células B de crecimiento rápido no adyacente en estadio II, III y IV
Las opciones de tratamiento del linfoma no Hodgkin de células B de crecimiento rápido no adyacente en estadio II, III y IV son las siguientes:
- Polatuzumab vedotina, rituximab, ciclofosfamida, doxorrubicina y prednisona.
- Rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona.
- Otra quimioterapia combinada.
- Consolidación con radioterapia dirigida a los sitios de enfermedad con gran masa tumoral (en evaluación clínica).
Polatuzumab vedotina, rituximab, ciclofosfamida, doxorrubicina y prednisona
Se comparó el régimen R-CHOP con el régimen de polatuzumab vedotina, rituximab, ciclofosfamida, doxorrubicina y prednisona (Pola-R-CHP). El polatuzumab es un conjugado anticuerpo-fármaco compuesto por un anticuerpo monoclonal anti-CD79B unido a la vedotina (monometil auristatina E), un inhibidor de los microtúbulos.
Evidencia (polatuzumab vedotina, rituximab, ciclofosfamida, doxorrubicina y prednisona):
- En un estudio prospectivo aleatorizado de 879 pacientes con LDCBG sin tratamiento previo, se compararon los regímenes R-CHOP y polatuzumab vedotina, rituximab, ciclofosfamida, doxorrubicina y prednisona (Pola-R-CHP). El polatuzumab vedotina se sustituyó por la vincristina para mitigar la toxicidad neurológica.
- Al cabo de una mediana de seguimiento de 28,2 meses, la tasa de supervivencia sin progresión (SSP) a 2 años fue significativamente más alta en el grupo de Pola-R-CHP que en el grupo de R-CHOP: 76,7 % (intervalo de confianza [IC] 95 %, 72,7–80,0 %) versus 70,2 % (IC 95 %, 65,8–74,6 %) (cociente de riesgos instantáneos [CRI], 0,73; IC 95 %, 0,57–0,95; P = 0,02).[Nivel de evidencia B1]
- La tasa de supervivencia general (SG) a 2 años fue del 88,7 % (IC 95 %, 85,7–91,6 %) para los pacientes que recibieron Pola-R-CHP y del 88,6 % (IC 95 %, 85,6–91,6 %) para los pacientes que recibieron R-CHOP (CRI, 0,94; IC 95 %, 0,65–1,37; P = 0,75).
- En un análisis de subpoblación asiática de este ensayo, se observó una mejora similar del 7,7 % en la SSP a 2 años con Pola-R-CHP versus R-CHOP.[Nivel de evidencia C2]
El intervalo de seguimiento fue demasiado corto en esta publicación para establecer si la mejora del 6 % en la tasa de SSP se estabilizará o mejorará después de 2 años, y no hay evidencia de ventaja en la SG. No obstante, los desenlaces actualizados con una mediana de seguimiento durante 3 años mostraron una mejora continua de la SSP, lo que llevó a la aprobación de este régimen por parte de la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA). El nuevo régimen cuesta más del doble que R-CHOP según los precios de adquisición de 2022, y es posible que el polatuzumab no esté disponible en muchos entornos del mundo entero.
No hay estudios validados para la intensificación del tratamiento de acuerdo con la tomografía por emisión de positrones interina. El R-CHOP tiene potencial curativo incluso en pacientes mayores de 80 años que son frágiles y requieren dosis reducidas de los componentes del R-CHOP. En una revisión retrospectiva de 239 pacientes, la tasa de supervivencia por causa específica a 5 años fue del 48 % (IC 95 %, 41−55 %).[Nivel de evidencia C3]
Menos del 10 % de los pacientes con LDCBG presentan un linfoma de crecimiento lento simultáneo en el momento del diagnóstico, y en su mayoría son del fenotipo de célula B de centro germinativo. En una revisión retrospectiva de 1324 pacientes se observaron una supervivencia sin complicaciones (SSC) (CRI, 1,19) y una SG (CRI, 1,09) similares.[Nivel de evidencia C3] En 847 pacientes tratados con R-CHOP y que no presentaban enfermedad 24 meses después del tratamiento, la tasa de recaída del linfoma de crecimiento lento a los 5 años fue más alta con un diagnóstico de linfoma folicular simultáneo (7,4 vs. 2,1 % a los 5 años, P < 0,01) y con un fenotipo de células B de centro germinativo (3,9 vs. 0,0 % a los 5 años, P = 0,02).
Rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona
En los siguientes estudios, se estableció el régimen de rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona (R-CHOP) como un régimen estándar para los pacientes con linfoma difuso de células B grandes (LDCBG) recién diagnosticado y enfermedad no adyacente en estadio II, estadio III y estadio IV durante más de 20 años. La intensificación de la dosis de R-CHOP en un ciclo de 14 días versus un ciclo de 21 días no produjo mejores desenlaces. R-CHOP es el régimen preferido cuando el polatuzumab no está disponible o no es asequible, o cuando está contraindicado debido a efectos secundarios adversos. El régimen Pola-R-CHP es el primero en más de 20 años en ser aprobado por la FDA para el tratamiento de pacientes con enfermedad no adyacente en estadio II, estadio III y estadio IV.
Evidencia (rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona):
- El régimen de rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona (R-CHOP) mostró una mejora de la SSC y la SG en comparación con el régimen de ciclofosfamida, doxorrubicina, vincristina y prednisona (CHOP) solo, en 399 pacientes con LDCBG en estadio avanzado mayores de 60 años (tasa de SSC, 57 vs. 38 %; P = 0,002, y tasa de SG, 70 vs. 57 %;P = 0,007 a los 2 años).[Nivel de evidencia A1] Al cabo de una mediana de seguimiento de 10 años, la tasa de SG de los pacientes que recibieron R-CHOP en comparación con los pacientes que recibieron CHOP fue del 44 % versus el 28 % (P < 0,0001).
- De manera similar, en 326 pacientes evaluables menores de 61 años, el régimen R-CHOP mostró una mejora de la SSC y la SG en comparación con CHOP solo (tasa de SSC, 79 vs. 59 %, P = 0,001, y tasa de SG, 93 vs. 84 %,P = 0,001 a los 3 años).[Nivel de evidencia A1]
- En un estudio aleatorizado (DSHNHL-1999-1A [NCT00052936]) de 1222 pacientes mayores de 60 años, se comparó el régimen R-CHOP administrado cada 2 semanas durante 6 u 8 ciclos con el régimen CHOP administrado cada 2 semanas durante 6 u 8 ciclos. Después de una mediana de seguimiento de 72 meses, la SSC favoreció la administración de R-CHOP cada 2 semanas durante 6 u 8 ciclos (tasa de SSC a 6 años, 74 vs. 56 %; P < 0,0001). La SG favoreció el régimen R-CHOP por solo 6 ciclos debido al aumento de efectos tóxicos en el grupo de 8 ciclos (tasa de SG a 6 años, 90 vs. 80 %; P = 0,0004).[Nivel de evidencia A1] No se hizo una comparación entre los regímenes estándar de R-CHOP o CHOP administrados cada 3 semanas.
- En un ensayo (NCT00140595) de 380 pacientes menores de 60 años con LDCBG y una puntuación de 1 del International Prognostic Index (IPI) ajustada por edad, los pacientes se asignaron de manera aleatoria para recibir ACVBP y R-ACVBP más consolidación con metotrexato, ifosfamida, etopósido y citarabina versus CHOP y rituximab. Después de una mediana de seguimiento de 44 meses, las tasas de SG a 3 años favorecieron al régimen R-ACVBP (92 vs. 84 %; cociente de riesgos instantáneos [CRI], 0,44; IC 95 %, 0,28–0,81,P = 0,007).[Nivel de evidencia A1] Los efectos tóxicos considerablemente peores con R-ACVBP, la población destinataria reducida (<60 años con concentración elevada de lactato–deshidrogenasa [LDH] o enfermedad en estadio III o IV, pero no ambas) y la falta de un ensayo confirmatorio quizás impidan la adopción del régimen R-ACVBP como un nuevo estándar de atención.
En los ensayos clínicos, se siguen explorando modificaciones del régimen R-CHOP para mejorar la eficacia.
Consolidación con radioterapia dirigida a los sitios de enfermedad con gran masa tumoral
Después de la quimioterapia de inducción con R-CHOP (o regímenes similares), la adición de radioterapia de campo comprometido dirigida a los sitios de enfermedad con gran masa tumoral inicial (≥5–10 cm) o a sitios extralinfáticos aún es polémica. Se debe considerar el aumento de los riesgos, como la aparición de efectos tóxicos a largo plazo (por ejemplo, neoplasias malignas secundarias).
Trasplante de médula ósea o trasplante de células madre
En varios ensayos prospectivos aleatorizados se evaluó la función de la consolidación con trasplante de médula ósea (TMO) autógeno o trasplante de células madre (TCM) autógeno versus la quimioterapia sola en pacientes con linfoma difuso de células grandes en la primera remisión.; [Nivel de evidencia A1] Aunque en algunos de estos ensayos se observaron aumentos significativos de la SSC (del 10 % al 20 %) en los pacientes que recibieron terapia de dosis altas, no se pudieron demostrar diferencias significativas en la SG de forma prospectiva en ninguna de las series.
En análisis retrospectivos con pacientes de riesgo intermedio (2 factores de riesgo) o riesgo alto (más de 3 factores de riesgo) según el puntaje de IPI, se indicó una mejora para la supervivencia con el TMO en 2 ensayos. En estos estudios no se establece que la consolidación con dosis altas tiene valor para los pacientes con linfoma de crecimiento rápido que tienen un riesgo alto real de recaída; además, se demuestra que la SSC quizás es un sustituto inadecuado de la SG en estos pacientes.
Linfoma difuso de células B grandes gástrico en estadio IE o IIE
En 4 series de casos de más de 100 pacientes con enfermedad en estadio IE o IIE (con tejido linfático asociado a mucosas o sin este) y con infección por Helicobacter pylori, se notificó que más del 50 % de los pacientes alcanzaron una remisión completa duradera después de un tratamiento adecuado con antibióticos para erradicar el H. pylori.[Nivel de evidencia C3]
Factores pronósticos
En el índice pronóstico internacional de la National Comprehensive Cancer Network (National Comprehensive Cancer Network International Prognostic Index [NCCN-IPI]) para el linfoma no Hodgkin (LNH) de crecimiento rápido (linfoma difuso de células grandes) se identifican los siguientes 5 factores de riesgo significativos para el pronóstico de la supervivencia general (SG) y los puntajes de riesgo relacionado:
- Edad.
- <40 años: 0.
- 41–60 años: 1.
- 61–75 años: 2.
- >75 años: 3.
- Estadio III o IV: 1.
- Estado funcional (EF) 2, 3 o 4: 1.
- Concentración sérica de lactato–deshidrogenasa (LDH).
- Normalizada: 0.
- >1x–3x: 1.
- >3x: 2.
- Número de sitios extraganglionares ≥2: 1.
Puntajes de riesgo:
- Bajo (0 o 1): Tasa de SG a 5 años, 96 %; tasa de supervivencia sin progresión (SSP), 91 %.
- Intermedio bajo (2 o 3): Tasa de SG a 5 años, 82 %; tasa de SSP, 74 %.
- Intermedio alto (4 o 5): Tasa de SG a 5 años, 64 %; tasa de SSP, 51 %.
- Alto (>6): Tasa de SG a 5 años, 33 %; tasa de SSP, 30 %.
Se utilizan modificaciones del IPI ajustadas por edad y estadio para los pacientes más jóvenes con enfermedad localizada. Los intervalos de tiempo más cortos entre el diagnóstico y el tratamiento son marcadores indirectos de factores biológicos de pronóstico precario.
La identificación del gen BCL2 y el reordenamiento del gen MYC o la sobreexpresión dual del gen MYC, o ambos, confieren un pronóstico muy adverso. Los pacientes en riesgo alto de recaída se consideran aptos para participar en ensayos clínicos. La obtención de perfiles moleculares de expresión génica mediante micromatrices de ADN quizás sirva en el futuro para ayudar a estratificar a los pacientes en grupos de tratamiento dirigido a dianas específicas y para mejorar la predicción de la supervivencia después de la quimioterapia estándar.
Tratamiento del síndrome de lisis tumoral
Los pacientes con linfadenopatía extensa, gran masa tumoral, elevación del ácido úrico sérico y la LDH tienen un riesgo alto de síndrome de lisis tumoral que da lugar a trastornos metabólicos como hiperuricemia, hiperpotasemia, hiperfosfatemia, hipocalcemia e insuficiencia renal aguda subsiguiente. Las opciones de tratamiento son hidratación alcalina, alopurinol y rasburicasa, una urato–oxidasa recombinante.
Profilaxis del sistema nervioso central
El sistema nervioso central (CNS)-IPI es un instrumento usado para predecir qué pacientes tienen un riesgo mayor al 10 % de recaída en el sistema nervioso central (SNC). El German Lymphoma Study Group formuló este instrumento y se validó en la base de datos de British Columbia Cancer Agency. Se usó la presencia de 4 a 6 de los factores de riesgo del CNS-IPI (edad >60 años, estado funcional ≥2, concentración elevada de LDH, enfermedad en estadio III o IV, >1 sitio extraganglionar o compromiso renal o suprarrenal) para definir el grupo de riesgo alto para la recaída en el SNC (un riesgo del 12 % de compromiso del SNC al cabo de 2 años).
Por lo general, se recomienda la profilaxis del SNC (a menudo con 4 a 6 dosis de metotrexato intratecal) para los pacientes con compromiso testicular.[Nivel de evidencia C3] En un análisis retrospectivo de los estudios alemanes RICOVER, se comparó el metotrexato intratecal con la ausencia de profilaxis en los pacientes con LDCBG. Este estudio se llevó a cabo durante la era del tratamiento con R-CHOP. Con la posible excepción de los pacientes con compromiso testicular, el análisis mostró que el metotrexato intratecal no disminuyó el riesgo de enfermedad en el SNC.[Nivel de evidencia C3] Algunos profesionales clínicos emplean dosis altas intravenosas (IV) (a menudo 4 dosis) de metotrexato en lugar del tratamiento intratecal porque esto mejora la administración del fármaco y reduce la morbilidad del paciente. En 5 estudios retrospectivos y en 1 metanálisis en red se evaluó el metotrexato de dosis altas en monoterapia en pacientes con LDCBG de riesgo alto, y tampoco se observó mejora en la tasa de recaída en el SNC.[Nivel de evidencia C3] Los pacientes que se consideran de riesgo alto de recaída en el SNC (por ejemplo, los pacientes con enfermedad testicular, renal o suprarrenal, y 3 o más sitios extraganglionares) pueden recibir metotrexato intratecal o metotrexato de dosis altas IV, pero la falta de estudios aleatorizados confirmatorios pone en duda este estándar y muestra una necesidad urgente de mejores tratamientos verificados en ensayos clínicos. Aunque no hay evidencia suficiente que respalde un beneficio significativo de la profilaxis del SNC en la mayoría de los pacientes de riesgo alto, el riesgo percibido de no tratar la recaída del SNC a menudo ha superado la falta de evidencia de su eficacia. Aunque los pacientes con compromiso testicular, renal o suprarrenal se consideran una excepción, ya que solo hay un beneficio anecdótico con el metotrexato intratecal o el metotrexato de dosis altas IV en la reducción de la recidiva en el SNC.[Nivel de evidencia C3]
En análisis retrospectivos, la adición de rituximab a los regímenes a base de CHOP redujo de modo significativo el riesgo de recaída en el SNC.[Nivel de evidencia C3] Los pacientes con diseminación al SNC en el momento del diagnóstico o en el momento de la recaída suelen recibir rituximab y metotrexato de dosis altas o citarabina seguidas de trasplante de células madre (TCM) autógeno, pero este abordaje no se ha evaluado en ensayos aleatorizados.[Nivel de evidencia C3]
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
References
- Shankland KR, Armitage JO, Hancock BW: Non-Hodgkin lymphoma. Lancet 380 (9844): 848-57, 2012.
- Tilly H, Morschhauser F, Sehn LH, et al.: Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N Engl J Med 386 (4): 351-363, 2022.
- Song Y, Tilly H, Rai S, et al.: Polatuzumab vedotin in previously untreated DLBCL: an Asia subpopulation analysis from the phase 3 POLARIX trial. Blood 141 (16): 1971-1981, 2023.
- Dührsen U, Müller S, Hertenstein B, et al.: Positron Emission Tomography-Guided Therapy of Aggressive Non-Hodgkin Lymphomas (PETAL): A Multicenter, Randomized Phase III Trial. J Clin Oncol 36 (20): 2024-2034, 2018.
- Gobba S, Moccia AA, Gulden-Sala W, et al.: Outcome of patients older than 80 years with diffuse large B-cell lymphoma (DLBCL) treated with "standard" immunochemotherapy: A large retrospective study from 4 institutions. Hematol Oncol 36 (1): 84-92, 2018.
- Wang Y, Link BK, Witzig TE, et al.: Impact of concurrent indolent lymphoma on the clinical outcome of newly diagnosed diffuse large B-cell lymphoma. Blood 134 (16): 1289-1297, 2019.
- Wang Y, Farooq U, Link BK, et al.: Late Relapses in Patients With Diffuse Large B-Cell Lymphoma Treated With Immunochemotherapy. J Clin Oncol 37 (21): 1819-1827, 2019.
- Coiffier B: State-of-the-art therapeutics: diffuse large B-cell lymphoma. J Clin Oncol 23 (26): 6387-93, 2005.
- Cunningham D, Hawkes EA, Jack A, et al.: Rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone in patients with newly diagnosed diffuse large B-cell non-Hodgkin lymphoma: a phase 3 comparison of dose intensification with 14-day versus 21-day cycles. Lancet 381 (9880): 1817-26, 2013.
- Coiffier B, Lepage E, Briere J, et al.: CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 346 (4): 235-42, 2002.
- Coiffier B, Thieblemont C, Van Den Neste E, et al.: Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d'Etudes des Lymphomes de l'Adulte. Blood 116 (12): 2040-5, 2010.
- Pfreundschuh M, Trümper L, Osterborg A, et al.: CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol 7 (5): 379-91, 2006.
- Pfreundschuh M, Kuhnt E, Trümper L, et al.: CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol 12 (11): 1013-22, 2011.
- Récher C, Coiffier B, Haioun C, et al.: Intensified chemotherapy with ACVBP plus rituximab versus standard CHOP plus rituximab for the treatment of diffuse large B-cell lymphoma (LNH03-2B): an open-label randomised phase 3 trial. Lancet 378 (9806): 1858-67, 2011.
- Casasnovas RO, Ysebaert L, Thieblemont C, et al.: FDG-PET-driven consolidation strategy in diffuse large B-cell lymphoma: final results of a randomized phase 2 study. Blood 130 (11): 1315-1326, 2017.
- Held G, Murawski N, Ziepert M, et al.: Role of radiotherapy to bulky disease in elderly patients with aggressive B-cell lymphoma. J Clin Oncol 32 (11): 1112-8, 2014.
- Kahl BS: Bulky aggressive B-cell lymphoma: to radiate or not to radiate--that is the question. J Clin Oncol 32 (11): 1097-8, 2014.
- Phan J, Mazloom A, Medeiros LJ, et al.: Benefit of consolidative radiation therapy in patients with diffuse large B-cell lymphoma treated with R-CHOP chemotherapy. J Clin Oncol 28 (27): 4170-6, 2010.
- Haioun C, Lepage E, Gisselbrecht C, et al.: Survival benefit of high-dose therapy in poor-risk aggressive non-Hodgkin's lymphoma: final analysis of the prospective LNH87-2 protocol--a groupe d'Etude des lymphomes de l'Adulte study. J Clin Oncol 18 (16): 3025-30, 2000.
- Haioun C, Lepage E, Gisselbrecht C, et al.: Benefit of autologous bone marrow transplantation over sequential chemotherapy in poor-risk aggressive non-Hodgkin's lymphoma: updated results of the prospective study LNH87-2. Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 15 (3): 1131-7, 1997.
- Santini G, Salvagno L, Leoni P, et al.: VACOP-B versus VACOP-B plus autologous bone marrow transplantation for advanced diffuse non-Hodgkin's lymphoma: results of a prospective randomized trial by the non-Hodgkin's Lymphoma Cooperative Study Group. J Clin Oncol 16 (8): 2796-802, 1998.
- Gianni AM, Bregni M, Siena S, et al.: High-dose chemotherapy and autologous bone marrow transplantation compared with MACOP-B in aggressive B-cell lymphoma. N Engl J Med 336 (18): 1290-7, 1997.
- Kluin-Nelemans HC, Zagonel V, Anastasopoulou A, et al.: Standard chemotherapy with or without high-dose chemotherapy for aggressive non-Hodgkin's lymphoma: randomized phase III EORTC study. J Natl Cancer Inst 93 (1): 22-30, 2001.
- Gisselbrecht C, Lepage E, Molina T, et al.: Shortened first-line high-dose chemotherapy for patients with poor-prognosis aggressive lymphoma. J Clin Oncol 20 (10): 2472-9, 2002.
- Martelli M, Gherlinzoni F, De Renzo A, et al.: Early autologous stem-cell transplantation versus conventional chemotherapy as front-line therapy in high-risk, aggressive non-Hodgkin's lymphoma: an Italian multicenter randomized trial. J Clin Oncol 21 (7): 1255-62, 2003.
- Milpied N, Deconinck E, Gaillard F, et al.: Initial treatment of aggressive lymphoma with high-dose chemotherapy and autologous stem-cell support. N Engl J Med 350 (13): 1287-95, 2004.
- Betticher DC, Martinelli G, Radford JA, et al.: Sequential high dose chemotherapy as initial treatment for aggressive sub-types of non-Hodgkin lymphoma: results of the international randomized phase III trial (MISTRAL). Ann Oncol 17 (10): 1546-52, 2006.
- Stiff PJ, Unger JM, Cook JR, et al.: Autologous transplantation as consolidation for aggressive non-Hodgkin's lymphoma. N Engl J Med 369 (18): 1681-90, 2013.
- Chiappella A, Martelli M, Angelucci E, et al.: Rituximab-dose-dense chemotherapy with or without high-dose chemotherapy plus autologous stem-cell transplantation in high-risk diffuse large B-cell lymphoma (DLCL04): final results of a multicentre, open-label, randomised, controlled, phase 3 study. Lancet Oncol 18 (8): 1076-1088, 2017.
- Shipp MA, Abeloff MD, Antman KH, et al.: International Consensus Conference on high-dose therapy with hematopoietic stem-cell transplantation in aggressive non-Hodgkin's lymphomas: report of the jury. Ann Oncol 10 (1): 13-9, 1999.
- Morgner A, Miehlke S, Fischbach W, et al.: Complete remission of primary high-grade B-cell gastric lymphoma after cure of Helicobacter pylori infection. J Clin Oncol 19 (7): 2041-8, 2001.
- Chen LT, Lin JT, Shyu RY, et al.: Prospective study of Helicobacter pylori eradication therapy in stage I(E) high-grade mucosa-associated lymphoid tissue lymphoma of the stomach. J Clin Oncol 19 (22): 4245-51, 2001.
- Chen LT, Lin JT, Tai JJ, et al.: Long-term results of anti-Helicobacter pylori therapy in early-stage gastric high-grade transformed MALT lymphoma. J Natl Cancer Inst 97 (18): 1345-53, 2005.
- Kuo SH, Yeh KH, Wu MS, et al.: Helicobacter pylori eradication therapy is effective in the treatment of early-stage H pylori-positive gastric diffuse large B-cell lymphomas. Blood 119 (21): 4838-44; quiz 5057, 2012.
- Zhou Z, Sehn LH, Rademaker AW, et al.: An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 123 (6): 837-42, 2014.
- Møller MB, Christensen BE, Pedersen NT: Prognosis of localized diffuse large B-cell lymphoma in younger patients. Cancer 98 (3): 516-21, 2003.
- Maurer MJ, Ghesquières H, Link BK, et al.: Diagnosis-to-Treatment Interval Is an Important Clinical Factor in Newly Diagnosed Diffuse Large B-Cell Lymphoma and Has Implication for Bias in Clinical Trials. J Clin Oncol 36 (16): 1603-1610, 2018.
- Cuccuini W, Briere J, Mounier N, et al.: MYC+ diffuse large B-cell lymphoma is not salvaged by classical R-ICE or R-DHAP followed by BEAM plus autologous stem cell transplantation. Blood 119 (20): 4619-24, 2012.
- Johnson NA, Slack GW, Savage KJ, et al.: Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 30 (28): 3452-9, 2012.
- Green TM, Young KH, Visco C, et al.: Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 30 (28): 3460-7, 2012.
- Horn H, Ziepert M, Becher C, et al.: MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 121 (12): 2253-63, 2013.
- Canellos GP: CHOP may have been part of the beginning but certainly not the end: issues in risk-related therapy of large-cell lymphoma. J Clin Oncol 15 (5): 1713-6, 1997.
- Sha C, Barrans S, Cucco F, et al.: Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. J Clin Oncol 37 (3): 202-212, 2019.
- Coiffier B, Altman A, Pui CH, et al.: Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J Clin Oncol 26 (16): 2767-78, 2008.
- Cortes J, Moore JO, Maziarz RT, et al.: Control of plasma uric acid in adults at risk for tumor Lysis syndrome: efficacy and safety of rasburicase alone and rasburicase followed by allopurinol compared with allopurinol alone--results of a multicenter phase III study. J Clin Oncol 28 (27): 4207-13, 2010.
- Schmitz N, Zeynalova S, Nickelsen M, et al.: CNS International Prognostic Index: A Risk Model for CNS Relapse in Patients With Diffuse Large B-Cell Lymphoma Treated With R-CHOP. J Clin Oncol 34 (26): 3150-6, 2016.
- Zucca E, Conconi A, Mughal TI, et al.: Patterns of outcome and prognostic factors in primary large-cell lymphoma of the testis in a survey by the International Extranodal Lymphoma Study Group. J Clin Oncol 21 (1): 20-7, 2003.
- Vitolo U, Chiappella A, Ferreri AJ, et al.: First-line treatment for primary testicular diffuse large B-cell lymphoma with rituximab-CHOP, CNS prophylaxis, and contralateral testis irradiation: final results of an international phase II trial. J Clin Oncol 29 (20): 2766-72, 2011.
- Cheah CY, Wirth A, Seymour JF: Primary testicular lymphoma. Blood 123 (4): 486-93, 2014.
- Boehme V, Schmitz N, Zeynalova S, et al.: CNS events in elderly patients with aggressive lymphoma treated with modern chemotherapy (CHOP-14) with or without rituximab: an analysis of patients treated in the RICOVER-60 trial of the German High-Grade Non-Hodgkin Lymphoma Study Group (DSHNHL). Blood 113 (17): 3896-902, 2009.
- Glantz MJ, Cole BF, Recht L, et al.: High-dose intravenous methotrexate for patients with nonleukemic leptomeningeal cancer: is intrathecal chemotherapy necessary? J Clin Oncol 16 (4): 1561-7, 1998.
- Puckrin R, El Darsa H, Ghosh S, et al.: Ineffectiveness of high-dose methotrexate for prevention of CNS relapse in diffuse large B-cell lymphoma. Am J Hematol 96 (7): 764-771, 2021.
- Jeong H, Cho H, Kim H, et al.: Efficacy and safety of prophylactic high-dose MTX in high-risk DLBCL: a treatment intent-based analysis. Blood Adv 5 (8): 2142-2152, 2021.
- Orellana-Noia VM, Reed DR, McCook AA, et al.: Single-route CNS prophylaxis for aggressive non-Hodgkin lymphomas: real-world outcomes from 21 US academic institutions. Blood 139 (3): 413-423, 2022.
- Lewis KL, Jakobsen LH, Villa D, et al.: High-dose methotrexate is not associated with reduction in CNS relapse in patients with aggressive B-cell lymphoma: an international retrospective study of 2300 high-risk patients. [Abstract] Blood 138 (Suppl 1): A-181, 2021.
- Wilson MR, Eyre TA, Kirkwood AA, et al.: Timing of high-dose methotrexate CNS prophylaxis in DLBCL: a multicenter international analysis of 1384 patients. Blood 139 (16): 2499-2511, 2022.
- Haddad PA, Hammoud D, Gallagher KM: Effective central nervous system (CNS) prophylaxis chemotherapy approach in high risk diffuse large B-cell lymphoma (DLBCL): a network meta-analysis. [Abstract] Blood 138 (Suppl 1): A-1461, 2021.
- Eyre TA, Savage KJ, Cheah CY, et al.: CNS prophylaxis for diffuse large B-cell lymphoma. Lancet Oncol 23 (9): e416-e426, 2022.
- Villa D, Connors JM, Shenkier TN, et al.: Incidence and risk factors for central nervous system relapse in patients with diffuse large B-cell lymphoma: the impact of the addition of rituximab to CHOP chemotherapy. Ann Oncol 21 (5): 1046-52, 2010.
- Ferreri AJ, Donadoni G, Cabras MG, et al.: High Doses of Antimetabolites Followed by High-Dose Sequential Chemoimmunotherapy and Autologous Stem-Cell Transplantation in Patients With Systemic B-Cell Lymphoma and Secondary CNS Involvement: Final Results of a Multicenter Phase II Trial. J Clin Oncol 33 (33): 3903-10, 2015.
- Schmitz N, Wu HS: Advances in the Treatment of Secondary CNS Lymphoma. J Clin Oncol 33 (33): 3851-3, 2015.
Tratamiento del linfoma no Hodgkin de células B de crecimiento rápido recidivante
Opciones de tratamiento del linfoma no Hodgkin de células B de crecimiento rápido recidivante
En una revisión retrospectiva de múltiples ensayos internacionales, se identificaron 636 pacientes con linfoma difuso de células B grandes (LDCBG) resistente al tratamiento que se definió como enfermedad progresiva o estable, durante o justo en el momento de finalizar un ciclo de quimioterapia completo, o la recaída dentro del primer año posterior a un trasplante de células madre (TCM) autógeno. Cuando se usó terapia subsiguiente, la tasa de respuesta objetiva fue del 26 %, la tasa de respuesta completa (RC) fue del 7 %, la mediana de supervivencia general (SG) fue de 6,3 meses y solo el 20 % de los pacientes estaban vivos a los 2 años. Incluso con la quimioterapia de reinducción con un TCM autógeno planificado, los desenlaces siguen siendo precarios.
Las opciones de tratamiento del linfoma no Hodgkin de células B de crecimiento rápido recidivante son las siguientes:
- Terapia de células T con receptor de antígeno quimérico para la enfermedad primaria resistente al tratamiento o en recaída dentro de 1 año.
- Consolidación con trasplante de médula ósea o células madre.
- Terapia de células T con CAR para la recaída después de un trasplante de células madre autógeno.
- Tafasitamab y lenalidomida.
- Rituximab y lenalidomida.
- Polatuzumab vedotina con rituximab y bendamustina.
- Loncastuximab tesirina.
- Captadores biespecíficos de células T.
- Radioterapia paliativa.
Terapia de células T con receptor de antígeno quimérico para la enfermedad primaria resistente al tratamiento o en recaída dentro de 1 año
Los pacientes con LDCB que presentan recaída durante el tratamiento o dentro de los 2 meses siguientes a recibir la quimioterapia con rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona (R-CHOP) tienen enfermedad primaria resistente al tratamiento. Cualquier paciente con recaída de la enfermedad en el plazo de 1 año después de la quimioterapia con R-CHOP o con enfermedad primaria resistente al tratamiento tiene un pronóstico precario incluso con la reinducción mediante quimioinmunoterapia seguida de un TCM autógeno. Los pacientes que recibieron terapia de células T con CAR tuvieron una tasa de supervivencia sin progresión (SSP) a 3 años del 40 % al 50 % al cabo de un seguimiento de 40 meses, un resultado equivalente de manera retrospectiva al del TCM autógeno en los registros de médula ósea.
En 3 ensayos aleatorizados, se comparó la quimioinmunoterapia seguida de un TCM autógeno y terapia de células T con CAR con quimioinmunoterapia puente o sin esta para pacientes con riesgo alto de recaída de la enfermedad, definida como enfermedad primaria resistente al tratamiento o en recaída dentro de los 12 meses posteriores al tratamiento con R-CHOP inicial.
Evidencia (terapia de células T con CAR):
- En un ensayo aleatorizado prospectivo se incluyeron 359 pacientes con enfermedad primaria resistente al tratamiento o en recaída dentro de los 12 meses posteriores a la quimioterapia R-CHOP inicial. Los pacientes recibieron la terapia de células T con CAR, axicabtagén ciloleucel (axi-cel), con solo corticoesteroides como terapia puente o quimioinmunoterapia de segunda línea (por lo general rituximab, ifosfamida, etopósido y carboplatino [R-ICE] o rituximab, dexametasona, citarabina de dosis altas y cisplatino [R-DHAP]) seguida de un TCM autógeno.
- Al cabo de una mediana de seguimiento de 47,2 meses, la mediana de SG no se alcanzó en la cohorte de axi-cel y fue de 31,1 meses en la cohorte de quimioinmunoterapia. La SG estimada a 4 años fue del 54,6 % para los pacientes que recibieron axi-cel y del 46,0 % para los pacientes que recibieron quimioinmunoterapia (cociente de riesgos instantáneos [CRI] de muerte, 0,73; intervalo de confianza [IC] 95 %, 0,54–0,98, P = 0,03).[Nivel de evidencia A1]
- La mediana de SSP evaluada por el investigador fue de 14,7 meses en la cohorte axi-cel y de 3,7 meses en la cohorte de quimioterapia (CRI, 0,51; IC 95 %, 0,38–0,67).
- En el grupo de quimioinmunoterapia, el 64 % de los pacientes nunca recibieron un TCM autógeno mientras estaban en el estudio debido a una respuesta inadecuada, progresión o muerte.
- Se obtuvieron diferencias clínica y estadísticamente significativas en la calidad de vida en el grupo de terapia de células T con CAR en los días 100 y 150, en comparación con el estándar de atención.[Nivel de evidencia A2]
- Se presentó síndrome de liberación de citocinas de grado 3 o 4 en el 6 % de los pacientes y neurotoxicidad de grado 3 o 4 en el 21 % de los pacientes.
- En un ensayo aleatorizado prospectivo se incluyeron 184 pacientes con enfermedad primaria resistente al tratamiento o en recaída dentro de los 12 meses posteriores a la quimioterapia R-CHOP inicial. Los pacientes recibieron terapia de células T con CAR, lisocabtagén maraleucel; el 63 % de los pacientes recibieron terapia puente o quimioinmunoterapia de segunda línea, seguida de TCM autógeno.
- Al cabo de una mediana de seguimiento de 17,5 meses, no se alcanzó la mediana de SSP para los pacientes que recibieron lisocabtagén maraleucel y fue de 6,2 meses para los pacientes que recibieron quimioinmunoterapia seguida de TCM autógeno (CRI, 0,40; P < 0,0001).[Nivel de evidencia B1]
- En el grupo de quimioinmunoterapia del estudio, el 53 % de los pacientes nunca recibieron un TCM autógeno debido a una respuesta inadecuada, progresión o muerte.
- Se presentó síndrome de liberación de citocinas de grado 3 en el 1 % de los pacientes y neurotoxicidad de grado 3 en el 4 % de los pacientes. No se produjeron casos de grado 4 ni 5.
- En un ensayo aleatorizado prospectivo se incluyeron 322 pacientes con enfermedad primaria resistente al tratamiento o en recaída dentro de los 12 meses posteriores a la quimioterapia R-CHOP inicial. Los pacientes recibieron terapia de células T con CAR, tisagenlecleucel; la mayoría de los pacientes recibieron terapia puente para alcanzar una respuesta, o quimioinmunoterapia de segunda línea seguida de TCM autógeno.
- No hubo diferencia en la SSC para los pacientes de ninguno de los grupos (CRI, 1,07; IC 95 %, 0,82–1,40; P = 0,69).[Nivel de evidencia B1]
- En el grupo de terapia de células T con CAR, el 48 % de los pacientes recibieron dos o más ciclos de quimioinmunoterapia como parte de la terapia puente. Es posible que este abordaje de terapia puente haya producido un número inaceptable de casos de enfermedad progresiva.
En resumen:
- Para los pacientes con LDCBG recidivante de riesgo alto con enfermedad primaria resistente al tratamiento o en recaída dentro de los 12 meses posteriores al tratamiento con quimioterapia a base de R-CHOP, axi-cel y lisocabtagén maraleucel son regímenes de inducción superiores en comparación con los regímenes de quimioinmunoterapia, como R-ICE, R-DHAP y rituximab, gemcitabina, dexametasona y cisplatino (R-GDP).
- El intervalo hasta que los pacientes reciban las células T con CAR debe reducirse, la manera más óptima de lograr esto es mediante el uso de solo corticoesteroides, la eliminación de quimioinmunoterapia puente y la administración rápida (tanto como sea posible) de la infusión del producto de células T con CAR.
- La preferencia por la terapia de células T con CAR en lugar de la quimioinmunoterapia seguida de TCM autógeno no se aplica a los pacientes que recaen más de 12 meses después de la terapia con R-CHOP.
- La American Society of Clinical Oncology (ASCO) ha elaborado directrices para el tratamiento de los efectos adversos en pacientes que reciben terapia de células T con CAR.
Consolidación con trasplante de médula ósea o células madre
Trasplante de médula ósea
La consolidación con trasplante de médula ósea (TMO) es un tratamiento para los pacientes con linfoma en recaída. En estudios preliminares se indica que entre el 20 % y el 40 % de los pacientes permanecerán sin enfermedad a largo plazo, pero el porcentaje preciso depende de la selección de los pacientes y del tratamiento específico que se use. Los regímenes farmacológicos de preparación varían; algunos investigadores también usan la irradiación corporal total. Se logró un éxito similar con médula autógena, con médula purgada o sin purgar, y con médula alogénica.; []
Evidencia (trasplante de médula ósea):
- En un ensayo prospectivo aleatorizado (EORTC-PARMA) con 215 pacientes menores de 60 años en la primera o segunda recaída de un linfoma de crecimiento rápido y sin compromiso de la médula ósea o del sistema nervioso central, los pacientes recibieron 2 ciclos de quimioterapia combinada intensiva. Respondieron 109 pacientes que se asignaron de manera aleatoria para recibir 4 ciclos adicionales de quimioterapia y radioterapia de campo comprometido (RTCC) versus un trasplante de médula ósea (TMO) autógeno seguido de RTCC. Al cabo de una mediana de seguimiento a 5 años, la tasa de SSC mejoró significativamente con el trasplante (46 vs. 12 %). La tasa de SG también fue significativamente mejor con el trasplante (53 vs. 32 %).[Nivel de evidencia A1] El TMO de rescate no tuvo éxito en los pacientes con enfermedad en recaída del grupo sin trasplante.
En general, los pacientes que respondieron al tratamiento inicial y que respondieron al tratamiento convencional para la recaída antes del TMO obtuvieron los mejores resultados.
- En un ensayo prospectivo, los pacientes con recaída tardía (>12 meses después del diagnóstico) tuvieron mejor SG que los pacientes con recaída temprana (la tasa de supervivencia a 8 años fue del 29 vs. 13 %, P = 0,001).[Nivel de evidencia C1]
Trasplante de células madre periférico
El trasplante de células madre (TCM) periférico ha producido resultados equivalentes al TCM autógeno estándar. Incluso los pacientes que nunca presentaron una remisión completa con quimioterapia convencional pueden tener una SSP prolongada (31 % a los 5 años) después de la quimioterapia de dosis alta y un TCM hematopoyético si conservan la quimiosensibilidad a la terapia de reinducción.[Nivel de evidencia C2] Algunos pacientes con recaída después de un TCM autógeno previo pueden presentar remisiones duraderas después de un TCM alogénico mielosupresor o no mielosupresor.; [Nivel de evidencia C3] El acondicionamiento de intensidad reducida para el TCM alogénico suele incluir fludarabina con busulfano o fludarabina con ciclofosfamida, con 2 Gy de irradiación corporal total o sin este.
Evidencia (trasplante de células madre periférico):
- En un ensayo prospectivo aleatorizado, 396 pacientes con LDCBG en primera recaída o resistentes al tratamiento de primera línea recibieron R-ICE o R-DHAP seguidos de TCM autógeno.
- No hubo diferencia en la SSC o la SG a 3 años.[Nivel de evidencia A1]
- En un ensayo prospectivo aleatorizado, 619 pacientes con linfoma de crecimiento rápido en recaída o resistente al tratamiento recibieron R-DHAP o R-GDP, seguidos de TCM autógeno.
- Al cabo de una mediana de seguimiento de 53 meses, no hubo diferencias en la SSC o la SG, pero los pacientes que recibieron R-GDP notificaron menos efectos tóxicos.[Nivel de evidencia A3]
Terapia de células T con receptor de antígeno quimérico para la recaída después de un trasplante de células madre autógeno
En caso de recaída de la enfermedad después de un trasplante de células madre (TCM) autógeno, muchos pacientes reciben consolidación con terapia de células T con receptor de antígeno quimérico (células T con CAR).
En múltiples ensayos se describen pacientes con linfoma de células B grandes resistente al tratamiento que recibieron una infusión de células T modificadas para expresar un CAR dirigido al antígeno CD19 que se expresa en las células B malignas mediante el uso de 3 compuestos diferentes: axi-cel, tisagenlecleucel y lisocabtagén maraleucel. En cada estudio se notificó una tasa de RC del 50 % al 60 % y una tasa de SG a 2 años del 40 % al 50 %, pero aún no se ha determinado la duración a largo plazo de la respuesta en estos pacientes muy específicos.[Nivel de evidencia C3] Este tratamiento es una opción para los pacientes con enfermedad resistente al tratamiento. Estos resultados se verificaron fuera del estudio en 2 informes que incluyeron 397 pacientes que recibieron tratamiento después de la aprobación de la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA).[Nivel de evidencia C3] Es posible que los pacientes con riesgo más alto que responden bien reciban consolidación con un TCM alogénico subsecuente, en algunos casos si son aptos.
La ASCO ha elaborado directrices para el tratamiento de los efectos adversos en pacientes que reciben terapia de células T con CAR.
Tafasitamab y lenalidomida
Evidencia (tafasitamab y lenalidomida):
- En un estudio de fase II, 80 pacientes con LDCBG en recaída o resistente al tratamiento recibieron tafasitamab, un anticuerpo monoclonal humanizado dirigido a CD19 combinado con lenalidomida.
- La tasa de RC fue del 43 % y la tasa de respuesta objetiva fue del 60 %.
La FDA aprobó la combinación de tafasitamab y lenalidomida para pacientes con LDCBG en recaída o resistente al tratamiento.[Nivel de evidencia C3] No está claro si el uso de esta terapia dirigida a CD19 puede interferir en la consolidación con células T con CAR dirigida a CD19.
Rituximab y lenalidomida
Evidencia (rituximab y lenalidomida):
- En 2 ensayos de fase II, 49 pacientes demostraron una tasa de respuesta general para el rituximab y lenalidomida del 19 % al 35 %.[Nivel de evidencia C3]
Polatuzumab vedotina, rituximab y bendamustina
Polatuzumab vedotina es un anticuerpo monoclonal dirigido a CD79b conjugado con el citotóxico vedotina (un conjugado anticuerpo-fármaco).
Evidencia (polatuzumab vedotina, rituximab y bendamustina):
- En un ensayo aleatorizado prospectivo, se trataron 80 pacientes de LDCBG en recaída o resistente al tratamiento con polatuzumab vedotina, combinado con bendamustina y rituximab (BR) o BR solo; el criterio principal de valoración fue la RC.
- La tasa de RC determinada mediante tomografía por emisión de positrones y tomografía computarizada fue del 40 % para la combinación de polatuzumab vedotina y BR, en comparación con una tasa del 18 % para BR solo (P = 0,026).
- De manera similar, la mediana de la SSP fue más alta para los pacientes que recibieron la combinación de polatuzumab vedotina (9,5 meses) que para los pacientes que recibieron BR solo (3,7 meses) (CRI, 0,36; IC 95 %, 0,21−0,63; P < 0,001); la SG fue de 12,4 meses para los pacientes que recibieron la combinación de polatuzumab vedotina versus 4,7 meses para los pacientes que recibieron BR solo (CRI, 0,42; IC 95 %, 0,24−0,75; P = 0,002).[Nivel de evidencia C1]
La FDA aprobó la combinación de polatuzumab vedotina y BR para pacientes de LDCBG en recaída o resistente al tratamiento.
Loncastuximab tesirina
Loncastuximab tesirina es un anticuerpo dirigido a CD19 conjugado con una citotoxina del dímero pirrolobenzodiazepina (un conjugado anticuerpo-fármaco).
Evidencia (loncastuximab tesirina):
- En un ensayo de fase I y posterior de fase II, se incluyeron 184 pacientes con enfermedad en recaída o resistente al tratamiento.
- La tasa de respuesta general fue del 48,3 % (IC 95 %, 39,9–56,7 %) y la tasa de RC fue del 24 %.[Nivel de evidencia C3]
Captadores biespecíficos de células T
Los captadores biespecíficos de células T se unen a CD20 (o CD19) y a CD3 para dar instrucciones a las células T de eliminar o destruir los linfocitos B malignos. De manera similar a la terapia de células T con CAR, casi la mitad de los pacientes que reciben esta terapia experimentan el síndrome de liberación de citocinas.
Radioterapia paliativa
En general, los pacientes con linfoma de crecimiento rápido que recaen con un tipo histológico de crecimiento lento se benefician de la terapia paliativa. Es posible lograr la paliación con dosis muy bajas (4 Gy) de RTCC en los pacientes con recaída de la enfermedad de crecimiento rápido y lento.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
References
- Crump M, Neelapu SS, Farooq U, et al.: Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood 130 (16): 1800-1808, 2017.
- Ayers EC, Li S, Medeiros LJ, et al.: Outcomes in patients with aggressive B-cell non-Hodgkin lymphoma after intensive frontline treatment failure. Cancer 126 (2): 293-303, 2020.
- Schuster SJ, Tam CS, Borchmann P, et al.: Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol 22 (10): 1403-1415, 2021.
- Shah NN, Ahn KW, Litovich C, et al.: Is autologous transplant in relapsed DLBCL patients achieving only a PET+ PR appropriate in the CAR T-cell era? Blood 137 (10): 1416-1423, 2021.
- Shadman M, Pasquini M, Ahn KW, et al.: Autologous transplant vs chimeric antigen receptor T-cell therapy for relapsed DLBCL in partial remission. Blood 139 (9): 1330-1339, 2022.
- Cappell KM, Sherry RM, Yang JC, et al.: Long-Term Follow-Up of Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy. J Clin Oncol 38 (32): 3805-3815, 2020.
- Locke FL, Miklos DB, Jacobson CA, et al.: Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N Engl J Med 386 (7): 640-654, 2022.
- Westin JR, Oluwole OO, Kersten MJ, et al.: Survival with Axicabtagene Ciloleucel in Large B-Cell Lymphoma. N Engl J Med 389 (2): 148-157, 2023.
- Abramson JS, Solomon SR, Arnason J, et al.: Lisocabtagene maraleucel as second-line therapy for large B-cell lymphoma: primary analysis of the phase 3 TRANSFORM study. Blood 141 (14): 1675-1684, 2023.
- Bishop MR, Dickinson M, Purtill D, et al.: Second-Line Tisagenlecleucel or Standard Care in Aggressive B-Cell Lymphoma. N Engl J Med 386 (7): 629-639, 2022.
- Santomasso BD, Nastoupil LJ, Adkins S, et al.: Management of Immune-Related Adverse Events in Patients Treated With Chimeric Antigen Receptor T-Cell Therapy: ASCO Guideline. J Clin Oncol 39 (35): 3978-3992, 2021.
- Shipp MA, Abeloff MD, Antman KH, et al.: International Consensus Conference on high-dose therapy with hematopoietic stem-cell transplantation in aggressive non-Hodgkin's lymphomas: report of the jury. Ann Oncol 10 (1): 13-9, 1999.
- Freedman AS, Takvorian T, Anderson KC, et al.: Autologous bone marrow transplantation in B-cell non-Hodgkin's lymphoma: very low treatment-related mortality in 100 patients in sensitive relapse. J Clin Oncol 8 (5): 784-91, 1990.
- Phillips GL, Fay JW, Herzig RH, et al.: The treatment of progressive non-Hodgkin's lymphoma with intensive chemoradiotherapy and autologous marrow transplantation. Blood 75 (4): 831-8, 1990.
- Chopra R, Goldstone AH, Pearce R, et al.: Autologous versus allogeneic bone marrow transplantation for non-Hodgkin's lymphoma: a case-controlled analysis of the European Bone Marrow Transplant Group Registry data. J Clin Oncol 10 (11): 1690-5, 1992.
- Ratanatharathorn V, Uberti J, Karanes C, et al.: Prospective comparative trial of autologous versus allogeneic bone marrow transplantation in patients with non-Hodgkin's lymphoma. Blood 84 (4): 1050-5, 1994.
- Mills W, Chopra R, McMillan A, et al.: BEAM chemotherapy and autologous bone marrow transplantation for patients with relapsed or refractory non-Hodgkin's lymphoma. J Clin Oncol 13 (3): 588-95, 1995.
- Philip T, Guglielmi C, Hagenbeek A, et al.: Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin's lymphoma. N Engl J Med 333 (23): 1540-5, 1995.
- Vellenga E, van Putten WL, van 't Veer MB, et al.: Rituximab improves the treatment results of DHAP-VIM-DHAP and ASCT in relapsed/progressive aggressive CD20+ NHL: a prospective randomized HOVON trial. Blood 111 (2): 537-43, 2008.
- Guglielmi C, Gomez F, Philip T, et al.: Time to relapse has prognostic value in patients with aggressive lymphoma enrolled onto the Parma trial. J Clin Oncol 16 (10): 3264-9, 1998.
- Vose JM, Anderson JR, Kessinger A, et al.: High-dose chemotherapy and autologous hematopoietic stem-cell transplantation for aggressive non-Hodgkin's lymphoma. J Clin Oncol 11 (10): 1846-51, 1993.
- Liberti G, Pearce R, Taghipour G, et al.: Comparison of peripheral blood stem-cell and autologous bone marrow transplantation for lymphoma patients: a case-controlled analysis of the EBMT Registry data. Lymphoma Working Party of the EBMT. Ann Oncol 5 (Suppl 2): 151-3, 1994.
- Vose JM, Zhang MJ, Rowlings PA, et al.: Autologous transplantation for diffuse aggressive non-Hodgkin's lymphoma in patients never achieving remission: a report from the Autologous Blood and Marrow Transplant Registry. J Clin Oncol 19 (2): 406-13, 2001.
- van Kampen RJ, Canals C, Schouten HC, et al.: Allogeneic stem-cell transplantation as salvage therapy for patients with diffuse large B-cell non-Hodgkin's lymphoma relapsing after an autologous stem-cell transplantation: an analysis of the European Group for Blood and Marrow Transplantation Registry. J Clin Oncol 29 (10): 1342-8, 2011.
- Freytes CO, Loberiza FR, Rizzo JD, et al.: Myeloablative allogeneic hematopoietic stem cell transplantation in patients who experience relapse after autologous stem cell transplantation for lymphoma: a report of the International Bone Marrow Transplant Registry. Blood 104 (12): 3797-803, 2004.
- Rezvani AR, Norasetthada L, Gooley T, et al.: Non-myeloablative allogeneic haematopoietic cell transplantation for relapsed diffuse large B-cell lymphoma: a multicentre experience. Br J Haematol 143 (3): 395-403, 2008.
- Ghosh N, Ahmed S, Ahn KW, et al.: Association of Reduced-Intensity Conditioning Regimens With Overall Survival Among Patients With Non-Hodgkin Lymphoma Undergoing Allogeneic Transplant. JAMA Oncol 6 (7): 1011-1018, 2020.
- Gisselbrecht C, Glass B, Mounier N, et al.: Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol 28 (27): 4184-90, 2010.
- Crump M, Kuruvilla J, Couban S, et al.: Randomized comparison of gemcitabine, dexamethasone, and cisplatin versus dexamethasone, cytarabine, and cisplatin chemotherapy before autologous stem-cell transplantation for relapsed and refractory aggressive lymphomas: NCIC-CTG LY.12. J Clin Oncol 32 (31): 3490-6, 2014.
- Neelapu SS, Locke FL, Bartlett NL, et al.: Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 377 (26): 2531-2544, 2017.
- Schuster SJ, Bishop MR, Tam CS, et al.: Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 380 (1): 45-56, 2019.
- Locke FL, Ghobadi A, Jacobson CA, et al.: Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol 20 (1): 31-42, 2019.
- Abramson JS, Palomba ML, Gordon LI, et al.: Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 396 (10254): 839-852, 2020.
- Lin JK, Muffly LS, Spinner MA, et al.: Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Multiply Relapsed or Refractory Adult Large B-Cell Lymphoma. J Clin Oncol 37 (24): 2105-2119, 2019.
- Jacobson CA, Hunter BD, Redd R, et al.: Axicabtagene Ciloleucel in the Non-Trial Setting: Outcomes and Correlates of Response, Resistance, and Toxicity. J Clin Oncol 38 (27): 3095-3106, 2020.
- Nastoupil LJ, Jain MD, Feng L, et al.: Standard-of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results From the US Lymphoma CAR T Consortium. J Clin Oncol 38 (27): 3119-3128, 2020.
- Salles G, Duell J, González Barca E, et al.: Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol 21 (7): 978-988, 2020.
- Zinzani PL, Pellegrini C, Gandolfi L, et al.: Combination of lenalidomide and rituximab in elderly patients with relapsed or refractory diffuse large B-cell lymphoma: a phase 2 trial. Clin Lymphoma Myeloma Leuk 11 (6): 462-6, 2011.
- Wiernik PH, Lossos IS, Tuscano JM, et al.: Lenalidomide monotherapy in relapsed or refractory aggressive non-Hodgkin's lymphoma. J Clin Oncol 26 (30): 4952-7, 2008.
- Sehn LH, Herrera AF, Flowers CR, et al.: Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J Clin Oncol 38 (2): 155-165, 2020.
- Calabretta E, Hamadani M, Zinzani PL, et al.: The antibody-drug conjugate loncastuximab tesirine for the treatment of diffuse large B-cell lymphoma. Blood 140 (4): 303-308, 2022.
- Caimi PF, Ai W, Alderuccio JP, et al.: Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol 22 (6): 790-800, 2021.
- Hamadani M, Radford J, Carlo-Stella C, et al.: Final results of a phase 1 study of loncastuximab tesirine in relapsed/refractory B-cell non-Hodgkin lymphoma. Blood 137 (19): 2634-2645, 2021.
- Dickinson MJ, Carlo-Stella C, Morschhauser F, et al.: Glofitamab for Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 387 (24): 2220-2231, 2022.
- Lee AY, Connors JM, Klimo P, et al.: Late relapse in patients with diffuse large-cell lymphoma treated with MACOP-B. J Clin Oncol 15 (5): 1745-53, 1997.
- Haas RL, Poortmans P, de Jong D, et al.: Effective palliation by low dose local radiotherapy for recurrent and/or chemotherapy refractory non-follicular lymphoma patients. Eur J Cancer 41 (12): 1724-30, 2005.
Tratamiento del linfoma linfoblástico de células B y la leucemia linfocítica aguda de células B
El linfoma linfoblástico (LLB) es una forma muy maligna, es decir, de crecimiento rápido o agresiva, del linfoma no Hodgkin (LNH), que a menudo se presenta en pacientes jóvenes, pero no es exclusiva de este grupo de edad. También se llama linfoma linfoblástico de células precursoras. El LLB es la manifestación linfomatosa de la leucemia linfocítica aguda (LLA). Los paradigmas de tratamiento se basan en ensayos clínicos para la LLA, porque el LLB y la LLA se consideran diferentes manifestaciones de la misma enfermedad biológica. El LLB a menudo produce masas mediastínicas grandes y tiene una tendencia mucho más alta por la diseminación a la médula ósea y el sistema nervioso central (SNC). La quimioterapia combinada intensiva con profilaxis del SNC es el tratamiento estándar para este tipo histológico de crecimiento rápido de LNH. A veces, se administra radioterapia dirigida a las áreas con gran masa tumoral. Debido a que estas formas de LNH tienden a progresar rápido, la quimioterapia combinada se administra de inmediato una vez que se confirma el diagnóstico. Para obtener más información, consultar Tratamiento de la leucemia linfoblástica aguda en adultos.
Los aspectos más importantes de los exámenes para la estadificación pretratamiento incluyen una revisión cuidadosa de las siguientes muestras para análisis patológicos:
- Aspirado de médula ósea.
- Muestra de biopsia.
- Líquido cefalorraquídeo para análisis citológico.
- Marcadores de linfocitos.
Opciones de tratamiento del linfoma linfoblástico de células B y la leucemia linfocítica aguda de células B
Las opciones de tratamiento del linfoma linfoblástico de células B (LLB-B) son las siguientes:
- Tratamiento intensivo.
- Radioterapia.
Los grupos cooperativos nacionales están formulando abordajes nuevos de tratamiento. Otros abordajes incluyen la consolidación con trasplante de médula ósea.
Para obtener más información, consultar Tratamiento de la leucemia linfoblástica aguda en adultos.
Tratamiento intensivo
El tratamiento estándar es la quimioterapia combinada intensiva con profilaxis del SNC.
Radioterapia
A veces, se administra radioterapia dirigida a las áreas con gran masa tumoral.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
adult Burkitt lymphomaB-cell non-Hodgkin lymphomaTratamiento del linfoma difuso de células pequeñas no hendidas o linfoma de Burkitt
El linfoma difuso de células pequeñas no hendidas o linfoma de Burkitt, en general, afecta a pacientes más jóvenes y constituyen el tipo más común de linfoma no Hodgkin (LNH) infantil. El linfoma de células B de grado alto, sin otra indicación, incluye linfomas con características morfológicas tipo Burkitt o blastoides, sin mutación doble en el análisis citogenético y los que exhiben un fenotipo de células B de centro germinativo. Hasta la mitad de los pacientes presentan un único reordenamiento de MYC. El tratamiento óptimo no está bien definido porque el diagnóstico es poco frecuente. Se suelen elegir regímenes para el linfoma de Burkitt con profilaxis del sistema nervioso central (SNC).
Opciones de tratamiento del linfoma difuso de células pequeñas no hendidas o linfoma de Burkitt
Las opciones de tratamiento del linfoma difuso de células pequeñas no hendidas o linfoma de Burkitt son las siguientes:
- Regímenes multifarmacológicos intensivos.
- Profilaxis del sistema nervioso central.
Regímenes multifarmacológicos intensivos
El tratamiento del linfoma difuso de células pequeñas no hendidas o linfoma de Burkitt suele ser un régimen multifarmacológico intensivo similar a los que se usan para los linfomas de crecimiento rápido en estadio avanzado (como el difuso de células grandes). Los factores pronósticos adversos incluyen la edad mayor a 40 años, una elevación de las concentraciones séricas de lactato–deshidrogenasa (>3 veces de lo normal), estado funcional 2 o superior según el Eastern Cooperative Oncology Group y compromiso del SNC. En una revisión retrospectiva de 641 pacientes adultos con linfoma de Burkitt de 30 centros oncológicos de los Estados Unidos, se encontró una tasa de supervivencia sin progresión (SSP) a 3 años del 64 %. Entre los pacientes, el 19 % presentó compromiso en el SNC y el 14 % presentó enfermedad primaria resistente al tratamiento; la tasa de mortalidad relacionada con el tratamiento fue del 10 %.
Evidencia (regímenes multifarmacológicos intensivos):
- La quimioterapia combinada intensiva, similar a la que se usa para el linfoma de Burkitt infantil, resultó muy exitosa en pacientes adultos. Más del 60 % de los pacientes en estadio avanzado estaban sin enfermedad a los 5 años.
- El rituximab se ha incorporado a estos regímenes intensivos de quimioterapia combinada. En un ensayo no aleatorizado multicéntrico prospectivo de un solo grupo, en 363 pacientes de 16 a 85 años, se observó una tasa de SSP a 5 años del 71 % y una tasa de supervivencia general a 5 años del 80 %.[Nivel de evidencia C1]
Profilaxis del sistema nervioso central
Los pacientes con linfoma difuso de células pequeñas no hendidas o linfoma de Burkitt tienen un riesgo de por vida del 20 % al 30 % de compromiso del SNC. Se recomienda la profilaxis del SNC con metotrexato para todos los pacientes, por lo general se administran de 4 a 6 inyecciones intratecales. Para obtener más información, consultar Tratamiento de la leucemia linfoblástica aguda en adultos.
Evidencia (profilaxis del sistema nervioso central):
- En una serie de 41 pacientes tratados con quimioterapia sistémica e intratecal, el 44 % de aquellos que presentaron enfermedad en el SNC y el 13 % de aquellos que recayeron con compromiso del SNC sobrevivieron a largo plazo sin enfermedad. Los modelos de recaída en el SNC fueron similares con independencia de que los pacientes recibieran o no radioterapia, pero se notó un aumento de alteraciones neurológicas en aquellos que recibieron radioterapia.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
References
- Blum KA, Lozanski G, Byrd JC: Adult Burkitt leukemia and lymphoma. Blood 104 (10): 3009-20, 2004.
- Evens AM, Danilov A, Jagadeesh D, et al.: Burkitt lymphoma in the modern era: real-world outcomes and prognostication across 30 US cancer centers. Blood 137 (3): 374-386, 2021.
- Olszewski AJ, Kurt H, Evens AM: Defining and treating high-grade B-cell lymphoma, NOS. Blood 140 (9): 943-954, 2022.
- Thomas DA, Faderl S, O'Brien S, et al.: Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer 106 (7): 1569-80, 2006.
- Hoelzer D, Walewski J, Döhner H, et al.: Improved outcome of adult Burkitt lymphoma/leukemia with rituximab and chemotherapy: report of a large prospective multicenter trial. Blood 124 (26): 3870-9, 2014.
- Roschewski M, Dunleavy K, Abramson JS, et al.: Multicenter Study of Risk-Adapted Therapy With Dose-Adjusted EPOCH-R in Adults With Untreated Burkitt Lymphoma. J Clin Oncol 38 (22): 2519-2529, 2020.
- Magrath I, Adde M, Shad A, et al.: Adults and children with small non-cleaved-cell lymphoma have a similar excellent outcome when treated with the same chemotherapy regimen. J Clin Oncol 14 (3): 925-34, 1996.
- Hoelzer D, Ludwig WD, Thiel E, et al.: Improved outcome in adult B-cell acute lymphoblastic leukemia. Blood 87 (2): 495-508, 1996.
- Mead GM, Sydes MR, Walewski J, et al.: An international evaluation of CODOX-M and CODOX-M alternating with IVAC in adult Burkitt's lymphoma: results of United Kingdom Lymphoma Group LY06 study. Ann Oncol 13 (8): 1264-74, 2002.
- Rizzieri DA, Johnson JL, Niedzwiecki D, et al.: Intensive chemotherapy with and without cranial radiation for Burkitt leukemia and lymphoma: final results of Cancer and Leukemia Group B Study 9251. Cancer 100 (7): 1438-48, 2004.
- Magrath IT, Haddy TB, Adde MA: Treatment of patients with high grade non-Hodgkin's lymphomas and central nervous system involvement: is radiation an essential component of therapy? Leuk Lymphoma 21 (1-2): 99-105, 1996.
Actualizaciones más recientes a este resumen (04/04/2024)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Información general sobre el linfoma no Hodgkin de células B
Se actualizaron las estadísticas con el número estimado de casos nuevos y defunciones para 2024 (se citó a la American Cancer Society como referencia 3).
Linfoma no Hodgkin de células B de crecimiento lento
Se añadió a Cartron et al. como referencia 25.
Se revisó el texto para indicar que los regímenes de primera línea para la macroglobulinemia de Waldenström incluyen zanubrutinib, rituximab e ibrutinib, rituximab solo, análogos nucleosídicos, y alquilantes, en monoterapia o quimioterapia combinada (se citó a Buske et al. como referencia 51). También se revisó el texto sobre los resultados de un ensayo en el que se comparó el zanubrutinib con el ibrutinib para indicar que, al cabo de una mediana de seguimiento de 44,4 meses, la tasa de supervivencia sin progresión (SSP) y la tasa de supervivencia general (SG) fueron similares en los dos grupos (se citó a Dimopoulos et al. como referencia 55).
Linfoma no Hodgkin de células B de crecimiento rápido
Se añadió texto para indicar que, aunque no hay evidencia suficiente que respalde un beneficio significativo de la profilaxis del SNC en la mayoría de los pacientes de riesgo alto, el riesgo percibido de no tratar la recaída en el SNC a menudo ha tenido más importancia que la falta de evidencia de su eficacia (se citó a Eyre et al. como referencia 34).
Se revisó el texto sobre los resultados de un ensayo de fase II sobre pembrolizumab en pacientes con linfoma mediastínico primario de células B grandes. Al cabo de una mediana de seguimiento de 48,7 meses, la tasa de SSP a 4 años fue del 33,0 % y la tasa de SG a 4 años del 45,3 % (se citó a Zinzani et al. como referencia 53 y nivel de referencia C3). También se añadió texto para indicar que los 11 pacientes que lograron una respuesta completa la seguían manteniendo en el momento del análisis final.
Se añadió texto para indicar que las mutaciones en TP53 se relacionan de manera especial con la progresión temprana del linfoma de células de manto (LCM) y con la muerte de los pacientes que reciben quimioinmunoterapia convencional e inhibidores de la tirosina–cinasa de Bruton (BTK) de segunda línea (se citó a Lew et al. como referencia 69).
Se revisó el texto para indicar que los ensayos aleatorizados no se han confirmado los beneficios para la supervivencia general (SG) en pacientes con LCM que reciben terapia de consolidación con TCM autógeno desde la introducción del rituximab. Se añadió texto para indicar que en un análisis retrospectivo de 1265 pacientes menores de 65 años aptos para trasplante no se observó ningún beneficio del TCM autógeno durante el tiempo que transcurrió hasta el siguiente tratamiento o SG. En este mismo análisis retrospectivo de cohortes del mundo real se halló un beneficio para el rituximab de mantenimiento durante el tiempo transcurrido hasta el siguiente tratamiento y la SG (se citó a Martin et al. como referencia 100 y nivel de referencia C3).
Se añadió texto sobre una evaluación retrospectiva posterior de 16 instituciones se incluyeron 168 pacientes que recibieron brexucabtagén autoleucel como parte del U.S. Lymphoma CAR-T Consortium. En el estudio se observaron resultados similares a los del ensayo ZUMA-2 (se citó a Wang Y et al. como referencia 107 y nivel de referencia C3).
Se añadió texto sobre un estudios de fase I/II de pirtobrutinib en 164 pacientes con LCM. La tasa de respuesta general fue del 57,8 %, incluida una tasa de respuesta completa del 20,0 %. La duración mediana de la respuesta fue de 21,6 meses (se citó a Wang ML et al. como referencia 108 y nivel de referencia C3). La Administración de Alimentos y Medicamentos de los Estados Unidos aprobó el uso de pirtobrutinib en pacientes que hayan recibido al menos 2 líneas de tratamiento previas, incluido otro inhibidor de la BTK.
Se revisó el texto para indicar que el rituximab, la lenalidomida, el ibrutinib, el acalabrutinib, el zanubrutinib, el pirtobrutinib y el venetoclax son fármacos biológicos directos que quizás conduzcan a estrategias de tratamiento sin quimioterapia para los pacientes con LCM. Se añadió texto para indicar que la mayoría de los estudios apoyan de 2 a 3 años de tratamiento de mantenimiento con rituximab tras la terapia de inducción (con o sin consolidación) (se citó a Forstpointner et al. como referencia 110).
Aspectos generales de las opciones de tratamiento del linfoma no Hodgkin de células B
Se revisó el Cuadro 4, Opciones de tratamiento del linfoma no Hodgkin de células B.
Tratamiento del linfoma no Hodgkin de células B de crecimiento lento en estadio I y del linfoma no Hodgkin de células B de crecimiento lento adyacente en estadio II
Se añadió a Cartron et al. como referencia 12.
Tratamiento del linfoma no Hodgkin de células B de crecimiento lento no adyacente en estadio II, III o IV
Se añadió a Cartron et al. como referencia 19.
Se añadió texto sobre los resultados de un ensayo prospectivo en el que participaron 202 pacientes con linfoma folicular de baja carga tumoral que no se trató previamente. Los pacientes se asignaron al azar a recibir 4 dosis semanales de rituximab intravenoso (IV) o una dosis de rituximab IV seguida de 3 dosis semanales de rituximab subcutáneo y dosis de mantenimiento en los meses 3, 5, 7 y 9 (se citó un nivel de referencia B1).
Tratamiento del linfoma no Hodgkin de células B de crecimiento rápido no adyacente en estadio II, III y IV
Se añadió texto para indicar que, aunque no hay evidencia suficiente que respalde un beneficio significativo de la profilaxis del SNC en la mayoría de los pacientes de riesgo alto, el riesgo percibido de no tratar la recaída del SNC a menudo ha superado la falta de evidencia de su eficacia (se citó a Eyre et al. como referencia 58).
El Consejo editorial del PDQ sobre el tratamiento para adultos es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este resumen del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento del linfoma no Hodgkin de células B. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El Consejo editorial del PDQ sobre el tratamiento para adultos, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
- Si el artículo se debe analizar en una reunión del consejo.
- Si conviene añadir texto acerca del artículo.
- Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Tratamiento del linfoma no Hodgkin de células B son:
- Eric J. Seifter, MD (Johns Hopkins University)
- Cole H. Sterling, MD (Johns Hopkins University)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El Consejo editorial del PDQ sobre el tratamiento para adultos emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como “En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]”.
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
PDQ® sobre el tratamiento para adultos. PDQ Tratamiento del linfoma no Hodgkin de células B. Bethesda, MD: National Cancer Institute. Actualización:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como “estándar” o “en evaluación clínica”. Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Comuníquese con el Instituto Nacional del Cáncer
Para obtener más información sobre las opciones para comunicarse con el NCI, incluso la dirección de correo electrónico, el número telefónico o el chat, consultar la página del Servicio de Información de Cáncer del Instituto Nacional del Cáncer.