Instituto Nacional del Cáncer
Fecha de publicación: Jan 22, 2024
Resumen de información revisada por expertos sobre el tratamiento del cáncer de tiroides.
Tratamiento del cáncer de tiroides infantil
Incidencia
La incidencia anual de cáncer de tiroides es de 4,8 a 5,9 casos por millón de personas de 0 a 19 años, lo que representa casi el 1,5 % de todos los cánceres en este grupo de edad. La incidencia de cáncer de tiroides es más alta en las personas de 15 a 19 años (17,6 casos por millón de personas) y representa casi el 8 % de todos los cánceres que se presentan en este grupo de mayor edad. Los carcinomas de tiroides se presentan con más frecuencia en las niñas que en los niños. La tendencia hacia tumores más grandes indica que el examen diagnóstico minucioso no es la única explicación de los resultados observados.
En 2 estudios de análisis de tendencia en el tiempo que usaron la base de datos Surveillance, Epidemiology, and End Results (SEER) Program, se observó un aumento del 2 % y el 3,8 % anual en la incidencia del carcinoma de tiroides diferenciado en niños, adolescentes y adultos jóvenes en los períodos entre 1973 y 2011, y 1984 y 2010, respectivamente. En Canadá, se ha documentado una tendencia general similar durante las últimas 2 décadas. En un estudio poblacional danés de cáncer de tiroides en pacientes menores de 24 años, se informó que la tasa de incidencia ajustada por edad (por 100 000) aumentó de manera significativa desde 0,36 en 1980 hasta 0,97 en 2014, con un promedio de cambio anual del 2,9 %. No se observaron cambios en la supervivencia general, pero sí un aumento importante de la incidencia de cáncer de tiroides en los adultos jóvenes (edad de 18–24 años), sobre todo en mujeres y en pacientes con carcinoma papilar.
La incidencia de cáncer de tiroides es más alta en personas blancas que en personas negras (5,3 casos por millón vs. 1,5 casos por millón). También es más alta en mujeres adolescentes que en varones adolescentes (8,1 casos por millón vs. 1,7 casos por millón).
El subtipo papilar es el cáncer de tiroides más común y representa alrededor del 60 % de los casos, seguido por el subtipo papilar de variante folicular (20–25 %), el subtipo folicular (10 %) y el subtipo medular (<10 %). La incidencia del subtipo papilar y su variante folicular llega a su punto máximo entre los 15 y los 19 años. La incidencia del cáncer de tiroides medular es la más alta en los niños de 0 a 4 años y disminuye a mayor edad (consultar la Figura 1).
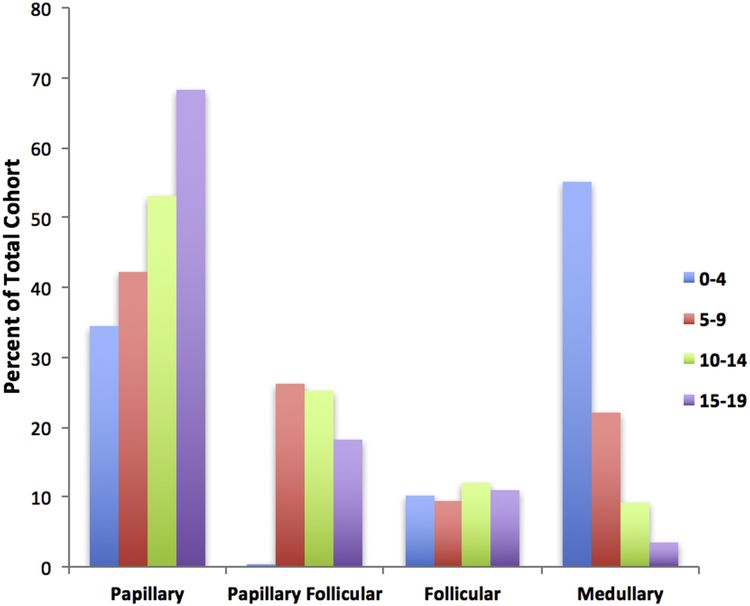 Figura 1. Incidencia del carcinoma de tiroides infantil según el subtipo más frecuente por 100 000 como porcentaje de la cohorte total. Reproducción autorizada de International Journal of Pediatric Otorhinolaryngology, Volume 89, Sarah Dermody, Andrew Walls, Earl H. Harley Jr., Pediatric thyroid cancer: An update from the SEER database 2007–2012, páginas 121–126, Derechos de autor (2016), con autorización de Elsevier. Percent of Total Cohort: porcentaje de la cohorte total; papillary: papilar; papillary follicular: papilar folicular; follicular: folicular; medullary: medular.
Figura 1. Incidencia del carcinoma de tiroides infantil según el subtipo más frecuente por 100 000 como porcentaje de la cohorte total. Reproducción autorizada de International Journal of Pediatric Otorhinolaryngology, Volume 89, Sarah Dermody, Andrew Walls, Earl H. Harley Jr., Pediatric thyroid cancer: An update from the SEER database 2007–2012, páginas 121–126, Derechos de autor (2016), con autorización de Elsevier. Percent of Total Cohort: porcentaje de la cohorte total; papillary: papilar; papillary follicular: papilar folicular; follicular: folicular; medullary: medular.
References
- Golpanian S, Perez EA, Tashiro J, et al.: Pediatric papillary thyroid carcinoma: outcomes and survival predictors in 2504 surgical patients. Pediatr Surg Int 32 (3): 201-8, 2016.
- Dermody S, Walls A, Harley EH: Pediatric thyroid cancer: An update from the SEER database 2007-2012. Int J Pediatr Otorhinolaryngol 89: 121-6, 2016.
- Horner MJ, Ries LA, Krapcho M, et al.: SEER Cancer Statistics Review, 1975-2006. National Cancer Institute, 2009. Also available online. Last accessed August 21, 2023.
- Shapiro NL, Bhattacharyya N: Population-based outcomes for pediatric thyroid carcinoma. Laryngoscope 115 (2): 337-40, 2005.
- Vergamini LB, Frazier AL, Abrantes FL, et al.: Increase in the incidence of differentiated thyroid carcinoma in children, adolescents, and young adults: a population-based study. J Pediatr 164 (6): 1481-5, 2014.
- Pole JD, Zuk AM, Wasserman JD: Diagnostic and Treatment Patterns Among Children, Adolescents, and Young Adults with Thyroid Cancer in Ontario: 1992-2010. Thyroid 27 (8): 1025-1033, 2017.
- Schmidt Jensen J, Grønhøj C, Mirian C, et al.: Incidence and Survival of Thyroid Cancer in Children, Adolescents, and Young Adults in Denmark: A Nationwide Study from 1980 to 2014. Thyroid 28 (9): 1128-1133, 2018.
Factores de riesgo
Los factores de riesgo para el cáncer de tiroides infantil son los siguientes:
- Exposición a radiación. Hay una frecuencia excesiva de adenomas y carcinomas de tiroides papilares después de la exposición a la radiación, debido a contaminación ambiental o por el uso de radiación ionizante para diagnóstico o tratamiento. El riesgo aumenta luego de exponerse a una dosis media de más de 0,05 a 0,1 Gy (50–100 mGy), sigue un patrón lineal dosis-respuesta hasta los 30 Gy y luego disminuye. El riesgo de cáncer de tiroides después de la exposición a radiación es aún mayor cuando se produce a menor edad y continúa durante más de 45 años después de haber ocurrido. Para obtener más información, consultar la sección Neoplasias subsiguientes en Efectos tardíos del tratamiento anticanceroso en la niñez.
El carcinoma de tiroides papilar es la forma más frecuente de carcinoma de tiroides que se diagnostica después de la exposición a radiación. Con frecuencia se encuentran alteraciones moleculares, que incluyen reordenamientos intracromosómicos; entre estos, los más comunes son los reordenamientos en RET/PTC.
- Nódulos tiroideos y tiroiditis autoinmunitaria. En un estudio de 485 nódulos en 385 niños que se sometieron a aspiración con aguja fina, se observó la presencia de cáncer de tiroides en 108 nódulos (24 %). Se observó tiroiditis autoinmunitaria en 95 pacientes (25 %) y se relacionó de forma independiente con un mayor riesgo de cáncer de tiroides (oportunidad relativa [OR], 2,19; intervalo de confianza 95 %, 1,32–3,62). El carcinoma de tiroides papilar fue más frecuente que el carcinoma de tiroides folicular. Entre los carcinomas de tiroides papilares, la tiroiditis autoinmunitaria estaba relacionada de manera importante con la variante esclerosante difusa (OR, 4,74).
- Herencia genética. La herencia genética es un factor importante en un subgrupo de carcinomas tiroideos. En los niños, la causa del carcinoma de tiroides medular es la mutación dominante de ganancia de función heredada o de novo en el protooncogén RET, asociada a la neoplasia endocrina múltiple (NEM) de tipo 2, ya sea NEM2A o NEM2B dependiendo de la mutación específica. En el caso de los pacientes con síndrome de NEM, el cáncer de tiroides puede estar asociado al desarrollo de otros tumores malignos. Para obtener más información, consultar Tratamiento de los síndromes de neoplasia endocrina múltiple infantiles.
- Antecedentes familiares. En el caso de los carcinomas de células foliculares, solo el 5 % al 10 % son cánceres familiares. Entre ellos, la mayoría de los casos familiares son asindrómicos, mientras que apenas una minoría ocurre en el contexto de síndromes de cáncer bien definidos con alteraciones de la línea germinal conocidas, como los siguientes:
- Poliposis asociada a APC.
- Complejo de Carney.
- Síndrome de tumores hamartomatosos relacionados con PTEN.
- Síndrome de Werner.
- Síndrome de DICER1.
References
- Cahoon EK, Nadyrov EA, Polyanskaya ON, et al.: Risk of Thyroid Nodules in Residents of Belarus Exposed to Chernobyl Fallout as Children and Adolescents. J Clin Endocrinol Metab 102 (7): 2207-2217, 2017.
- Rose J, Wertheim BC, Guerrero MA: Radiation treatment of patients with primary pediatric malignancies: risk of developing thyroid cancer as a secondary malignancy. Am J Surg 204 (6): 881-6; discussion 886-7, 2012.
- Lal G, Groff M, Howe JR, et al.: Risk of subsequent primary thyroid cancer after another malignancy: latency trends in a population-based study. Ann Surg Oncol 19 (6): 1887-96, 2012.
- Lubin JH, Adams MJ, Shore R, et al.: Thyroid Cancer Following Childhood Low-Dose Radiation Exposure: A Pooled Analysis of Nine Cohorts. J Clin Endocrinol Metab 102 (7): 2575-2583, 2017.
- Iglesias ML, Schmidt A, Ghuzlan AA, et al.: Radiation exposure and thyroid cancer: a review. Arch Endocrinol Metab 61 (2): 180-187, 2017 Mar-Apr.
- Keefe G, Culbreath K, Cherella CE, et al.: Autoimmune Thyroiditis and Risk of Malignancy in Children with Thyroid Nodules. Thyroid 32 (9): 1109-1117, 2022.
- Bauer AJ: Molecular Genetics of Thyroid Cancer in Children and Adolescents. Endocrinol Metab Clin North Am 46 (2): 389-403, 2017.
- Acquaviva G, Visani M, Repaci A, et al.: Molecular pathology of thyroid tumours of follicular cells: a review of genetic alterations and their clinicopathological relevance. Histopathology 72 (1): 6-31, 2018.
- Francis GL, Waguespack SG, Bauer AJ, et al.: Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 25 (7): 716-59, 2015.
Características histológicas
Los tumores de tiroides se clasifican como adenomas o carcinomas. Los adenomas son nódulos encapsulados benignos bien circunscritos que pueden agrandar parte o toda la glándula, la cual se extiende a ambos lados del cuello y llega a ser bastante grande. Algunos tumores segregan hormonas. Es posible que la transformación a un carcinoma maligno comience en algunas células que proliferan y diseminan a los ganglios linfáticos del cuello o los pulmones. Casi el 20 % de los nódulos tiroideos en los niños son malignos.
En la categoría general de diagnóstico de carcinoma de tiroides, se incluyen los siguientes tipos histológicos:
- Carcinoma de tiroides diferenciado. A menudo, se llama carcinomas de tiroides diferenciados a los carcinomas de tipo papilar y folicular. La clasificación patológica del carcinoma de tiroides diferenciado infantil, se basa en las definiciones estándar fijadas por la Organización Mundial de la Salud y los criterios son los mismos para niños y adultos. El desenlace clínico a largo plazo de los niños y adolescentes con carcinoma de tiroides diferenciado es excelente, con una tasa de supervivencia a 10 años superior al 95 %.
- Carcinoma de tiroides papilar. El carcinoma de tiroides papilar representa el 90 % o más de los casos de carcinoma de tiroides diferenciado que aparecen durante la infancia y la adolescencia. El carcinoma de tiroides papilar pediátrico se presenta con diferentes variantes histológicas: clásica, sólida, folicular y esclerosante difusa. El carcinoma de tiroides papilar es con frecuencia multifocal y bilateral, y metastatiza a los ganglios linfáticos regionales en la mayoría de los niños. En el 25 % de los casos se produce una metástasis hematógena.
- Cáncer de tiroides folicular. El cáncer de tiroides folicular es poco común. Por lo general, es un tumor unifocal y propenso a hacer metástasis hematógena inicial a los pulmones y los huesos. La metástasis a los ganglios linfáticos regionales es poco común. Las variantes histológicas del cáncer de tiroides folicular incluyen los carcinomas de células de Hürthle (oncocítico), el de células claras y el insular (poco diferenciado).
- Carcinoma de tiroides medular. El carcinoma de tiroides medular es una forma poco común de carcinoma de tiroides que se origina en las células C parafoliculares secretoras de calcitonina y se presenta en menos del 10 % de los casos de carcinoma de tiroides en niños. En niños con síndrome de neoplasia endocrina múltiple de tipo 2, el carcinoma de tiroides medular, por lo general, se relaciona con mutaciones de la línea germinal en RET.
- Carcinoma anaplásico. El carcinoma anaplásico representa menos del 1 % de los carcinomas de tiroides pediátricos.
References
- Dinauer C, Francis GL: Thyroid cancer in children. Endocrinol Metab Clin North Am 36 (3): 779-806, vii, 2007.
- Vasko V, Bauer AJ, Tuttle RM, et al.: Papillary and follicular thyroid cancers in children. Endocr Dev 10: 140-72, 2007.
- Halac I, Zimmerman D: Thyroid nodules and cancers in children. Endocrinol Metab Clin North Am 34 (3): 725-44, x, 2005.
- Francis GL, Waguespack SG, Bauer AJ, et al.: Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 25 (7): 716-59, 2015.
- Golpanian S, Perez EA, Tashiro J, et al.: Pediatric papillary thyroid carcinoma: outcomes and survival predictors in 2504 surgical patients. Pediatr Surg Int 32 (3): 201-8, 2016.
- Dermody S, Walls A, Harley EH: Pediatric thyroid cancer: An update from the SEER database 2007-2012. Int J Pediatr Otorhinolaryngol 89: 121-6, 2016.
- Brady C, Manning SC, Rudzinski E, et al.: Clinical Outcomes of Diffuse Sclerosing Variant Papillary Thyroid Carcinoma in Pediatric Patients. Laryngoscope 132 (5): 1132-1138, 2022.
- Sugino K, Nagahama M, Kitagawa W, et al.: Distant Metastasis in Pediatric and Adolescent Differentiated Thyroid Cancer: Clinical Outcomes and Risk Factor Analyses. J Clin Endocrinol Metab 105 (11): , 2020.
- Viola D, Romei C, Elisei R: Medullary thyroid carcinoma in children. Endocr Dev 26: 202-13, 2014.
Características moleculares
Carcinoma de tiroides de células foliculares
La oncogénesis de tiroides y la evolución de los carcinomas de células foliculares de tiroides (carcinoma de tiroides diferenciado, carcinoma de tiroides papilar poco diferenciado y carcinoma de tiroides anaplásico) se definen mediante un proceso de pasos múltiples que resulta en la activación anómala de las vías de señalización MAPK o PI3K/PTEN/AKT. Los estudios genómicos exhaustivos que se llevaron a cabo durante la última década han definido el panorama de estos tumores, así como también las correlaciones entre genotipo y fenotipo. Con el empleo de técnicas de secuenciación avanzada, se encuentran alteraciones oncógenas en más del 90 % de los tumores.
En una revisión retrospectiva se clasificaron 131 pacientes pediátricos con cáncer de tiroides diferenciado en 3 grupos: con mutaciones en RAS (HRAS, KRAS o NRAS), con mutaciones en BRAF (BRAF p.V600E) y con fusiones RET::NTRK (fusiones RET, NTRK1 y NTRK3). Los pacientes con fusiones RET::NTRK tenían una probabilidad significativamente mayor de presentar enfermedad avanzada en los ganglios linfáticos y metástasis a distancia y menos probabilidad de lograr la remisión al cabo de un año, en comparación con los pacientes de los subgrupos con mutaciones en RAS y mutaciones en BRAF.
Las mutaciones oncoiniciadoras más comunes son las que se presentan en los genes BRAF y RAS, seguidas de las fusiones de genes que comprometen RET o NTRK:
- BRAF. Las mutaciones puntuales en el gen BRAF son las alteraciones más comunes que se encuentran en el carcinoma de tiroides; la mutación más frecuente es la V600E (95 % de los casos de mutaciones en BRAF). Las mutaciones en BRAF se encuentran en el 40 % al 80 % de los carcinomas de tiroides papilares y en menor proporción en el carcinoma de tiroides papilar poco diferenciado (5–35 %) y en el carcinoma de tiroides anaplásico (10–50 %).
La presencia de BRAF V600E se relacionó con la extensión extratiroidea del tumor y con un aumento en el riesgo de recidiva; sin embargo, su importancia en el pronóstico es controvertida. Los tumores que presentan BRAF V600E parecen mostrar un perfil inmunosupresor amplio con expresión alta del ligando 1 de muerte celular no programada (PD-L1).
En un análisis retrospectivo de 80 pacientes brasileños menores de 18 años de edad con carcinoma de tiroides papilar se encontraron fusiones AGK::BRAF y mutaciones puntuales BRAF V600E. Las fusiones AGK::BRAF, que se encuentran en el 19 % de los pacientes pediátricos con carcinoma de tiroides papilar, se relacionaron con metástasis a distancia y menor edad. Las mutaciones BRAF V600E, que se encuentran en el 15 % de los pacientes con carcinoma de tiroides papilar pediátrico, se relacionaron con mayor edad y tumores más grandes.
- RAS. La activación oncogénica de RAS ocurre en cualquiera de las familias de genes RAS (NRAS, HRAS y KRAS) aunque las alteraciones más frecuentes son las mutaciones puntuales en NRAS. Las mutaciones en RAS son marcadores de lesiones de tiroides de patrón folicular. Estas aparecen en el 30 % al 50 % de los carcinomas de tiroides foliculares, en el 25 % al 45 % de las variantes foliculares del carcinoma de tiroides papilar y en menos del 10 % de los carcinomas de tiroides papilares. También se encuentran con frecuencia en el carcinoma de tiroides papilar poco diferenciado (20–50 %) y en el carcinoma de tiroides anaplásico (10–50 %) y se cree que promueven la progresión tumoral. Tienen una prevalencia mayor en las regiones con carencia de yodo.
- Reordenamientos en RET. Se han descubierto múltiples reordenamientos en RET en alrededor del 5 % al 25 % de los carcinomas de tiroides papilares y en menos del 10 % de sus variantes foliculares. Se encuentran muy vinculados a la exposición a la radiación ambiental. También son comunes en pacientes jóvenes, muchos de los cuales tienen metástasis ganglionares y manifestaciones clínicopatológicas de gran malignidad. Por último, se ha notificado que las mutaciones en RET son más frecuentes en la variante esclerosante difusa del carcinoma papilar que en el carcinoma papilar no esclerosante estándar (83 vs. 15,4 %; P = 0,0095).
- Reordenamientos en NTRK. Se han descrito los reordenamientos en NTRK1 y NTRK3 en alrededor del 5 % de los carcinomas de tiroides papilares; sin embargo, se informa sobre la presencia de ETV6::NTRK3 en el 15 % de los carcinomas de tiroides papilares provocados por radiación. En los pacientes jóvenes e infantiles, los carcinomas de tiroides papilares que tienen el reordenamiento en NTRK pueden presentar metástasis ganglionar y manifestaciones clinicopatológicas agresivas similares a las de los tumores con el reordenamiento en RET.
- Mutaciones en DICER1. Se encontraron alteraciones convencionales en 12 de 30 (40 %) carcinomas de tiroides papilar (5 casos con BRAF V600E, 3 casos con RET::CCDC6 y 4 casos con RET::NCOA4). Las mutaciones patógenas en el gen DICER1 se encontraron en cerca del 10 % de los carcinomas de tiroides papilar. También se encontraron mutaciones en DICER1 en una cohorte pequeña de pacientes con carcinomas de tiroides poco diferenciados.
En un estudio se correlacionó el estado de los puntos calientes de mutación en DICER1 con las características clínicas, histológicas y de desenlace en una serie de 56 pacientes pediátricos con carcinomas de tiroides papilares. Estos pacientes no tenían antecedentes clínicos ni familiares de manifestaciones sindrómicas relacionadas con DICER1. De los carcinomas de tiroides papilares, 15 (27%) albergaban la mutación BRAF p.V600E, mientras que 8 (14 %) presentaban mutaciones en DICER1, sin BRAF p.V600E asociadas. Se identificaron mutaciones en DICER1 en los exones 26 y 27. También se observó una nueva mutación D1810del (c.5428_5430delGAT). En el estudio se confirmó la ausencia de mutaciones en puntos calientes de DICER1 en el DNA del tejido no tumoral emparejado en los 8 carcinomas de tiroides papilares relacionados con DICER1. También se concluyó que la mayor incidencia en pacientes mujeres y el número elevado de carcinomas de tiroides papilares con patrón folicular de riesgo bajo son características de los carcinomas de tiroides papilares relacionados con DICER1.
Otras alteraciones incluyen las siguientes:
- Se han descrito reordenamientos en ALK en menos del 10 % de los carcinomas de tiroides papilares y en general se relacionan con desdiferenciaciones.
- Se han descrito mutaciones activadoras en AKT1 en menos del 19 % de los carcinomas de tiroides papilares poco diferenciados.
- Los reordenamientos en PPARG están presentes en el 20 % al 50 % de los carcinomas de tiroides foliculares y en menor proporción en las variantes foliculares del carcinoma de tiroides papilar.
- Normalmente se encuentran mutaciones activadoras en TERT en el carcinoma de tiroides papilar poco diferenciado (20–50 %) y en el carcinoma de tiroides anaplásico (30–75 %). Estas mutaciones también se notificaron en el 10 % al 35 % de los carcinomas de tiroides foliculares y en el 5 % al 15 % de los carcinomas de tiroides papilares. Se cree que las mutaciones en TERT promueven la progresión tumoral en los carcinomas de tiroides papilares poco diferenciados y carcinomas de tiroides anaplásicos, y representan un marcador de pronóstico negativo.
- TP53 está mutado en el 40 % al 80 % de los carcinomas de tiroides anaplásicos y en el 10 % al 35 % de los carcinomas de tiroides papilares poco diferenciados. Se considera que es el último paso en la progresión tumoral y un marcador de pronóstico poco favorable.
Al analizar los tumores con características histológicas similares, la gama de alteraciones genéticas difiere entre niños y adultos, en la siguiente forma:
- Las fusiones de genes que afectan a RET, o con menor frecuencia a NTRK, se presentan en alrededor del 50 % de las alteraciones moleculares en el carcinoma de tiroides infantil diferenciado, mientras que en los adultos se encuentran en cerca del 15 %.
- Las alteraciones en los genes BRAF o RAS, presentes en alrededor del 70 % de los carcinomas de tiroides que se diagnostican en adultos, se observan en el 20 % al 40 % de los tumores pediátricos; las mutaciones en BRAF se han descrito en cerca del 20 % al 30 % de los casos, mientras que las mutaciones en RAS se encuentran en niños con mucha menos frecuencia (5–10 %).
- Cuando se combinan las evaluaciones del DNA y RNA, es posible identificar alteraciones que quizás sirvan de diana en cerca del 98 % de los carcinomas de tiroides infantiles.
Carcinoma de tiroides medular
El carcinoma de tiroides medular es una neoplasia maligna neuroendocrina derivada de las células C parafoliculares de la glándula tiroides que se originan en la cresta neural. En niños, el carcinoma de tiroides medular es un desorden monogénico en el oncogén RET debido a una mutación dominante de ganancia de función heredada o de novo asociada a la neoplasia endocrina múltiple de tipo 2 (NEM2), ya sea NEM2A o NEM2B, dependiendo de la mutación específica. El riesgo más alto de carcinoma de tiroides medular lo confiere la mutación M918T en RET, que se asocia a NEM2B. Las mutaciones en RET asociadas a NEM2A confieren un riesgo menor de carcinoma de tiroides medular.
References
- Stosic A, Fuligni F, Anderson ND, et al.: Diverse Oncogenic Fusions and Distinct Gene Expression Patterns Define the Genomic Landscape of Pediatric Papillary Thyroid Carcinoma. Cancer Res 81 (22): 5625-5637, 2021.
- Franco AT, Ricarte-Filho JC, Isaza A, et al.: Fusion Oncogenes Are Associated With Increased Metastatic Capacity and Persistent Disease in Pediatric Thyroid Cancers. J Clin Oncol 40 (10): 1081-1090, 2022.
- Acquaviva G, Visani M, Repaci A, et al.: Molecular pathology of thyroid tumours of follicular cells: a review of genetic alterations and their clinicopathological relevance. Histopathology 72 (1): 6-31, 2018.
- Bauer AJ: Molecular Genetics of Thyroid Cancer in Children and Adolescents. Endocrinol Metab Clin North Am 46 (2): 389-403, 2017.
- Cancer Genome Atlas Research Network: Integrated genomic characterization of papillary thyroid carcinoma. Cell 159 (3): 676-90, 2014.
- Sisdelli L, Cordioli MICV, Vaisman F, et al.: AGK-BRAF is associated with distant metastasis and younger age in pediatric papillary thyroid carcinoma. Pediatr Blood Cancer 66 (7): e27707, 2019.
- Brady C, Manning SC, Rudzinski E, et al.: Clinical Outcomes of Diffuse Sclerosing Variant Papillary Thyroid Carcinoma in Pediatric Patients. Laryngoscope 132 (5): 1132-1138, 2022.
- Wasserman JD, Sabbaghian N, Fahiminiya S, et al.: DICER1 Mutations Are Frequent in Adolescent-Onset Papillary Thyroid Carcinoma. J Clin Endocrinol Metab 103 (5): 2009-2015, 2018.
- Chernock RD, Rivera B, Borrelli N, et al.: Poorly differentiated thyroid carcinoma of childhood and adolescence: a distinct entity characterized by DICER1 mutations. Mod Pathol 33 (7): 1264-1274, 2020.
- Onder S, Mete O, Yilmaz I, et al.: DICER1 Mutations Occur in More Than One-Third of Follicular-Patterned Pediatric Papillary Thyroid Carcinomas and Correlate with a Low-Risk Disease and Female Gender Predilection. Endocr Pathol 33 (4): 437-445, 2022.
- Potter SL, Reuther J, Chandramohan R, et al.: Integrated DNA and RNA sequencing reveals targetable alterations in metastatic pediatric papillary thyroid carcinoma. Pediatr Blood Cancer 68 (1): e28741, 2021.
- Pekova B, Sykorova V, Dvorakova S, et al.: RET, NTRK, ALK, BRAF, and MET Fusions in a Large Cohort of Pediatric Papillary Thyroid Carcinomas. Thyroid 30 (12): 1771-1780, 2020.
Cuadro clínico inicial y factores pronósticos
Carcinoma de tiroides diferenciado
Los pacientes de cáncer de tiroides por lo general presentan una masa tiroidea con adenopatía cervical indolora o sin ella. A partir de los antecedentes médicos y familiares, así como el conjunto de sus manifestaciones, el cáncer de tiroides quizá forme parte de un síndrome de predisposición tumoral como la neoplasia endocrina múltiple (NEM), la poliposis asociada a APC, el síndrome hamartomatoso PTEN, el complejo de Carney, el síndrome de Werner o el síndrome DICER1.
La edad más joven se relaciona con una presentación clínica más maligna del carcinoma de tiroides diferenciado. Se dieron a conocer las siguientes observaciones:
- En un estudio transversal que incluyó el 20 % de los hospitales comunitarios en los Estados Unidos, se comparó la presentación del cuadro clínico inicial de 644 pacientes pediátricos con más de 43 000 casos de adultos. Los niños, comparados con los adultos, tuvieron una proporción más alta de compromiso ganglionar (31,5 % en niños vs. 14,7 % en adultos) y metástasis pulmonares (5,7 % en niños vs. 2,2 % en adultos).
- Las tasas de recidiva más altas se vincularon con la presentación del cuadro clínico inicial a edades más jóvenes.
- Los tumores de mayor tamaño (>1 cm), la diseminación extratiroidea y la enfermedad multifocal se relacionan con un aumento del riesgo de metástasis ganglionares.
- Los niños prepuberales presentan un cuadro clínico inicial más maligno en comparación con los adolescentes en etapa puberal; además, los niños tienen un grado más alto de diseminación extratiroidea, compromiso de ganglios linfáticos y metástasis pulmonares. Sin embargo, el desenlace es similar entre los grupos de niños prepuberales y adolescentes.
- En un estudio de 106 pacientes pediátricos con carcinoma de tiroides papilar, en 80 pacientes se identificaron alteraciones genómicas. De estos 80 pacientes, 47 tenían mutaciones BRAFV600E, 31 tenían oncogenes de fusión (21 con RET, 6 con ALK y 4 con NTRK), 5 tenían variantes de DICER1, y 2 tenían una amplificación de FGFR y EGFR. Los pacientes con tumores positivos para una fusión eran más jóvenes (edad <10 años, 93 %); tenían una proporción más alta de tumores grandes (>2 cm), extensión extratiroidea, metástasis ganglionares y pulmonares, además de mayor incidencia de enfermedad recidivante o persistente en comparación con los pacientes de tumores con mutaciones en BRAF. La expresión de SLC5A5 (codificador de la proteína cotransportadora de sodio y yodo, un determinante importante de la avidez por I 131 [131I]) se redujo en los niños con carcinomas de tiroides papilares positivos para fusiones y en 2 pacientes con enfermedad resistente a 131I que albergaban una fusión de NTRK y de RET, respectivamente. La administración de larotrectinib y selpercatinib produjo respuestas tumorales y restauró la captación de yodo radioactivo, lo que señala la importancia de las pruebas moleculares en pacientes pediátricos con cáncer de tiroides papilar.
- En un ensayo se informaron los desenlaces de 65 pacientes chinos (edad <20 años) con carcinoma de tiroides papilar y metástasis pulmonares. De los pacientes, 20 presentaron metástasis pulmonares persistentes después del tratamiento con yodo radioactivo, a lo que se denominó enfermedad resistente al yodo radioactivo (RAIR). No se observó diferencia significativa en las características patológicas entre los pacientes menores de 15 años y los pacientes de 15 a 20 años, pero los más jóvenes tuvieron una probabilidad más alta de presentar la enfermedad RAIR (cociente de riesgos instantáneos [CRI], 3,500; intervalo de confianza [IC] 95 %, 1,134–10,803; P = 0,023). La enfermedad RAIR se identificó como un factor de predicción independiente de enfermedad progresiva (CRI, 10,008; IC 95 %, 2,427–41,268; P = 0,001). En la curva Kaplan-Meier se observó que el grupo RAIR tenía tasas más bajas de supervivencia sin progresión (SSP) y de supervivencia por una enfermedad específica que el grupo ávido de yodo radioactivo (P< 0,001 y P = 0,039). De forma similar, la enfermedad RAIR fue un factor de riesgo para una SSP desfavorable en pacientes menores de 15 años (P< 0,001).
En el caso del cáncer de tiroides bien diferenciado, se encontró que el sexo masculino, un tamaño grande del tumor y las metástasis a distancia tienen importancia pronóstica de mortalidad temprana; sin embargo, incluso los pacientes del grupo de riesgo más alto con metástasis a distancia tuvieron una tasa de supervivencia del 90 %. En un análisis de un registro francés, se observaron desenlaces similares en niños y adultos jóvenes que presentaron carcinoma de tiroides papilar después de recibir radioterapia en comparación con niños y adultos jóvenes con carcinoma de tiroides papilar espontáneo. Sin embargo, los pacientes que recibieron antes irradiación tiroidea para una enfermedad benigna padecieron más tumores invasores y compromiso ganglionar.
Una revisión de la National Cancer Database encontró que los pacientes de 21 años o menos de familias de bajos recursos y aquellos que no tenían seguro médico, tuvieron un mayor tiempo de espera entre el diagnóstico y el tratamiento del cáncer de tiroides bien diferenciado y presentaron un estadio más avanzado de la enfermedad.
Carcinoma de tiroides medular
Los niños con carcinoma de tiroides medular poseen un cuadro clínico más maligno; el 50 % de los casos presentan metástasis hematógenas en el momento del diagnóstico. El Instituto Nacional del Cáncer está realizando un estudio sobre la evolución natural del cáncer de tiroides medular en niños y adultos jóvenes (NCT01660984). En una revisión de 430 pacientes de 0 a 21 años con cáncer de tiroides medular, se notificó que la edad más avanzada (16–21 años) en el momento del diagnóstico, un tumor de más de 2 cm de diámetro, los márgenes afectados por cáncer después de una tiroidectomía total y las metástasis ganglionares se relacionaban con un pronóstico menos favorable.
Entre 1997 y 2019, en el registro de la German Society for Pediatric Oncology and Hematology–Malignant Endocrine Tumors se identificó a un total de 57 pacientes de carcinoma de tiroides medular y a 17 pacientes con hiperplasia de células C.[Nivel de evidencia C1] Para los pacientes de carcinoma de tiroides medular, la mediana de seguimiento fue de 5 años (intervalo, 0–19 años) y la mediana de edad en el momento del diagnóstico fue de 10 años (intervalo, 0–17 años). La tasa de supervivencia general fue del 87 % y la tasa de supervivencia sin complicaciones (SSC) fue del 52 %. En total, los síndromes de NEM de tipo 2 (NEM2) afectaron al 96,4 % de los pacientes; 37 de 42 exhibían NEM2A y 3 de 28 pacientes exhibían NEM2B (mutación M918T en RET). La tasa de SSC a 10 años fue del 78 % para los pacientes con NEM2A y del 38 % para los pacientes con NEM2B (P< 0,001). En análisis multivariantes, el estado de los ganglios linfáticos y las concentraciones altas de calcitonina después de la cirugía fueron importantes factores pronósticos adversos para la SSC.
En niños con NEM2B hereditaria, el carcinoma de tiroides medular a veces se detecta durante el primer año de vida y es posible que ocurran metástasis ganglionares antes de los 5 años de edad. El reconocimiento de neuromas mucosos, antecedentes de alacrimia, estreñimiento (secundario a ganglioneuromatosis) y características faciales y hábito marfanoide es crítico para el diagnóstico temprano porque la mutación RET M918T, asociada a NEM2B, es muchas veces una mutación de novo. Alrededor del 50 % de los pacientes con NEM2B desarrollan un feocromocitoma, mientras que con NEM2A el grado de riesgo de presentar un feocromocitoma e hipertiroidismo es variable según la mutación específica en RET. Para obtener más información, consultar Tratamiento de los síndromes de neoplasia endocrina múltiple infantiles.
References
- Al-Qurayshi Z, Hauch A, Srivastav S, et al.: A National Perspective of the Risk, Presentation, and Outcomes of Pediatric Thyroid Cancer. JAMA Otolaryngol Head Neck Surg 142 (5): 472-8, 2016.
- Acquaviva G, Visani M, Repaci A, et al.: Molecular pathology of thyroid tumours of follicular cells: a review of genetic alterations and their clinicopathological relevance. Histopathology 72 (1): 6-31, 2018.
- Francis GL, Waguespack SG, Bauer AJ, et al.: Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 25 (7): 716-59, 2015.
- Ye B, Shi J, Shen C, et al.: Comparison of differentiated thyroid carcinoma recurrence and its clinical features in children of different ages. Oncotarget 8 (29): 48051-48059, 2017.
- Kim J, Sun Z, Adam MA, et al.: Predictors of nodal metastasis in pediatric differentiated thyroid cancer. J Pediatr Surg 52 (1): 120-123, 2017.
- Lazar L, Lebenthal Y, Steinmetz A, et al.: Differentiated thyroid carcinoma in pediatric patients: comparison of presentation and course between pre-pubertal children and adolescents. J Pediatr 154 (5): 708-14, 2009.
- Redlich A, Luster M, Lorenz K, et al.: Age, American Thyroid Association Risk Group, and Response to Therapy Are Prognostic Factors in Children With Differentiated Thyroid Cancer. J Clin Endocrinol Metab 107 (1): e165-e177, 2022.
- Chesover AD, Vali R, Hemmati SH, et al.: Lung Metastasis in Children with Differentiated Thyroid Cancer: Factors Associated with Diagnosis and Outcomes of Therapy. Thyroid 31 (1): 50-60, 2021.
- Lee YA, Lee H, Im SW, et al.: NTRK and RET fusion-directed therapy in pediatric thyroid cancer yields a tumor response and radioiodine uptake. J Clin Invest 131 (18): , 2021.
- Tian T, Huang S, Dai H, et al.: Radioactive Iodine-Refractory Pulmonary Metastases of Papillary Thyroid Cancer in Children, Adolescents, and Young Adults. J Clin Endocrinol Metab 108 (2): 306-314, 2023.
- Shayota BJ, Pawar SC, Chamberlain RS: MeSS: A novel prognostic scale specific for pediatric well-differentiated thyroid cancer: a population-based, SEER outcomes study. Surgery 154 (3): 429-35, 2013.
- Sassolas G, Hafdi-Nejjari Z, Casagranda L, et al.: Thyroid cancers in children, adolescents, and young adults with and without a history of childhood exposure to therapeutic radiation for other cancers. Thyroid 23 (7): 805-10, 2013.
- Garner EF, Maizlin II, Dellinger MB, et al.: Effects of socioeconomic status on children with well-differentiated thyroid cancer. Surgery 162 (3): 662-669, 2017.
- Waguespack SG, Rich TA, Perrier ND, et al.: Management of medullary thyroid carcinoma and MEN2 syndromes in childhood. Nat Rev Endocrinol 7 (10): 596-607, 2011.
- Raval MV, Sturgeon C, Bentrem DJ, et al.: Influence of lymph node metastases on survival in pediatric medullary thyroid cancer. J Pediatr Surg 45 (10): 1947-54, 2010.
- Kuhlen M, Frühwald MC, Dunstheimer DPA, et al.: Revisiting the genotype-phenotype correlation in children with medullary thyroid carcinoma: A report from the GPOH-MET registry. Pediatr Blood Cancer 67 (4): e28171, 2020.
- Bauer AJ: Molecular Genetics of Thyroid Cancer in Children and Adolescents. Endocrinol Metab Clin North Am 46 (2): 389-403, 2017.
- Wells SA, Asa SL, Dralle H, et al.: Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 25 (6): 567-610, 2015.
Evaluación diagnóstica
La evaluación inicial de un niño o adolescente con un nódulo tiroideo incluye los siguientes procedimientos:
- Ecografía de tiroides.
- Concentración sérica de la hormona estimulante de la tiroides (TSH).
- Concentración de la tiroglobulina sérica.
Las pruebas del funcionamiento de la tiroides suelen dar resultados normales, pero la tiroglobulina quizás esté elevada.
La aspiración con aguja fina como abordaje diagnóstico inicial es sensible y útil. Sin embargo, en casos dudosos se debe considerar realizar una biopsia o resección abiertas.
References
- Francis GL, Waguespack SG, Bauer AJ, et al.: Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 25 (7): 716-59, 2015.
Consideraciones especiales para el tratamiento de niños con cáncer
El cáncer en niños y adolescentes es infrecuente, aunque desde 1975 se ha observado un aumento gradual de la incidencia general. Se debe considerar la derivación a centros médicos que cuenten con un equipo multidisciplinario de especialistas en oncología con experiencia en el tratamiento de los cánceres que se presentan en la niñez y la adolescencia. Este equipo multidisciplinario incorpora la pericia de los siguientes profesionales de atención de la salud y otros para asegurar que los niños reciban el tratamiento, los cuidados médicos de apoyo y la rehabilitación que les permitan lograr una supervivencia y calidad de vida óptimas:
- Médicos de atención primaria.
- Cirujanos pediatras.
- Radioncólogos.
- Oncólogos o hematólogos pediatras.
- Especialistas en rehabilitación.
- Enfermeros especializados en pediatría.
- Trabajadores sociales.
- Profesionales de vida infantil.
- Psicólogos.
Para obtener información sobre los cuidados médicos de apoyo para niños y adolescentes con cáncer, consultar los resúmenes sobre Cuidados médicos de apoyo y cuidados paliativos.
La American Academy of Pediatrics estableció pautas para los centros de oncología pediátrica y su función en el tratamiento de los pacientes de cáncer infantil. En estos centros de oncología pediátrica, se dispone de ensayos clínicos para la mayoría de los tipos de cáncer que se presentan en niños y adolescentes, y se ofrece la oportunidad de participar a la mayoría de los pacientes y familiares. Por lo general, los ensayos clínicos para los niños y adolescentes con cáncer se diseñan a fin de comparar un tratamiento que parece mejor con el tratamiento estándar actual. La mayoría de los avances en la identificación de tratamientos curativos para los cánceres infantiles se lograron mediante ensayos clínicos. Para obtener información sobre los ensayos clínicos en curso, consultar el portal de Internet del NCI.
Se han logrado mejoras notables en la supervivencia de niños y adolescentes con cáncer. Entre 1975 y 2020, la mortalidad por cáncer infantil disminuyó en más del 50 %. Los niños y adolescentes sobrevivientes de cáncer necesitan un seguimiento minucioso, ya que es posible que los efectos secundarios del tratamiento del cáncer persistan o se presenten meses o años después de este. Para obtener más información sobre la incidencia, el tipo y la vigilancia de los efectos tardíos en los niños y adolescentes sobrevivientes de cáncer, consultar Efectos tardíos del tratamiento anticanceroso en la niñez.
El cáncer infantil es una enfermedad rara con cerca de 15 000 casos anuales diagnosticados antes de los 20 años de edad en los Estados Unidos. En la Rare Diseases Act of 2002 de los Estados Unidos, se define una enfermedad rara como la que afecta a poblaciones de menos de 200 000 personas. Por lo tanto, todos los cánceres infantiles se consideran enfermedades raras.
La designación de un tumor raro es diferente entre los grupos pediátricos y de adultos. En el caso de los adultos, se considera que un cáncer es raro cuando su incidencia anual es inferior a 6 casos por 100 000 personas. Representan hasta el 24 % de los cánceres diagnosticados en la Unión Europea y alrededor del 20 % de los cánceres diagnosticados en los Estados Unidos. Además, tal como se indica a continuación, la designación de un tumor infantil raro no es uniforme entre los grupos internacionales:
- En una iniciativa conjunta de la European Union Joint Action on Rare Cancers y el European Cooperative Study Group for Rare Pediatric Cancers se estimó que el 11 % de todos los cánceres en pacientes menores de 20 años se podrían clasificar como muy raros. Este grupo de consenso definió los cánceres muy raros como los cánceres con incidencia anual inferior a 2 casos por millón de personas. Sin embargo, también se incluyen en este grupo de tumores muy raros otros 3 tipos histológicos (carcinoma de tiroides, melanoma y cáncer de testículo) con incidencias superiores a 2 casos por millón de personas, porque se cuenta con poco conocimiento y experiencia sobre el tratamiento de estos tumores.
- El Children's Oncology Group (COG) define los cánceres raros en la niñez según la lista del subgrupo XI de la International Classification of Childhood Cancer, en la que se incluyen los cánceres de tiroides, los cánceres de piel melanoma y no melanoma, además de los múltiples tipos de carcinomas (por ejemplo, los carcinomas de corteza suprarrenal, los carcinomas de nasofaringe y la mayoría de los carcinomas de tipo adulto, como los cánceres de mama, los cánceres colorrectales, etc.). Estos cánceres representan casi el 5 % de aquellos diagnosticados en niños de 0 a 14 años y casi el 27 % de los que se diagnostican en adolescentes de 15 a 19 años.
La mayoría de los cánceres del subgrupo XI son melanomas o cánceres de tiroides, mientras que otros tipos de cáncer solo representan el 2 % de los cánceres en niños de 0 a 14 años y el 9,3 % de los cánceres en adolescentes de 15 a 19 años.
Estudiar estos cánceres raros es un reto por el número bajo de pacientes con cualquier diagnóstico individual, el predominio de estos cánceres raros en adolescentes y la carencia de ensayos clínicos con adolescentes que tienen estos cánceres.
También es posible obtener información sobre estos tumores en fuentes relacionadas con el cáncer en adultos, por ejemplo, el resumen Tratamiento del cáncer de tiroides.
References
- Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed December 15, 2023.
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 15, 2023.
- Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed August 18, 2023.
- Ward E, DeSantis C, Robbins A, et al.: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 64 (2): 83-103, 2014 Mar-Apr.
- Gatta G, Capocaccia R, Botta L, et al.: Burden and centralised treatment in Europe of rare tumours: results of RARECAREnet-a population-based study. Lancet Oncol 18 (8): 1022-1039, 2017.
- DeSantis CE, Kramer JL, Jemal A: The burden of rare cancers in the United States. CA Cancer J Clin 67 (4): 261-272, 2017.
- Ferrari A, Brecht IB, Gatta G, et al.: Defining and listing very rare cancers of paediatric age: consensus of the Joint Action on Rare Cancers in cooperation with the European Cooperative Study Group for Pediatric Rare Tumors. Eur J Cancer 110: 120-126, 2019.
- Pappo AS, Krailo M, Chen Z, et al.: Infrequent tumor initiative of the Children's Oncology Group: initial lessons learned and their impact on future plans. J Clin Oncol 28 (33): 5011-6, 2010.
Tratamiento del carcinoma de tiroides papilar y folicular
Las opciones de tratamiento del carcinoma de tiroides papilar y folicular (diferenciado) son las siguientes:
- Cirugía.
- Ablación con yodo radiactivo.
- Terapia dirigida.
En 2015, la American Thyroid Association (ATA) Task Force on Pediatric Thyroid Cancer publicó directrices para el tratamiento de los nódulos tiroideos y el cáncer de tiroides diferenciado en niños y adolescentes. Estas directrices (que se resumen a continuación) se basan en datos probatorios científicos y opiniones de paneles de expertos con una evaluación cuidadosa del grado de comprobación científica.
- Evaluación preoperatoria.
- Un ecografista con experiencia deberá obtener una ecografía detallada de todas las regiones del cuello usando una sonda de alta resolución y la técnica de Doppler. Se deberá realizar un examen ecográfico completo antes de la cirugía.
- Se debe considerar la adición de imágenes transversales (tomografía computarizada [TC] con contraste o imágenes por resonancia magnética) cuando se sospeche invasión del tracto aerodigestivo. Es importante destacar que, si se utilizan medios de contraste yodados, será necesario demorar la evaluación y el tratamiento con yodo radiactivo durante 2 a 3 meses hasta que disminuya la carga total de yodo en el cuerpo.
- Es posible considerar obtener imágenes del tórax (radiografía o TC) para pacientes con enfermedad considerable en los ganglios linfáticos cervicales.
- Se obtiene una centellografía nuclear de la tiroides solo si el paciente presenta al inicio inhibición de la hormona estimulante de la tiroides (TSH).
- No se recomienda el uso rutinario de una gammagrafía ósea ni de una tomografía por emisión de positrones (TEP) con flúor F18-fludesoxiglucosa.
- Cirugía.
Lo ideal es que la cirugía de tiroides en los niños la lleve a cabo un cirujano que tenga experiencia en la realización de procedimientos endocrinos en niños y en un hospital que cuente con toda la gama de especializaciones pediátricas.
- Tiroidectomía:
La tiroidectomía total es la opción de tratamiento recomendada para los pacientes con carcinoma papilar o folicular. La recomendación del panel de expertos de la ATA se basa en datos que indican un aumento de incidencia de enfermedad bilateral (30 %) y multifocal (65 %).
En pacientes con un tumor unilateral pequeño confinado en la glándula, se podría contemplar una tiroidectomía subtotal para limitar el daño permanente en esas estructuras; se deja una porción muy pequeña de tejido tiroideo (<1–2 %) en el punto de entrada del nervio laríngeo recurrente o de las glándulas paratiroideas superiores.
En un análisis retrospectivo se identificaron factores relacionados con la afectación bilateral de la tiroides en 115 pacientes pediátricos con cáncer de tiroides bien diferenciado. De 115 participantes, 47 (41 %) presentaron enfermedad bilateral. En el análisis multivariable, solo la multifocalidad en el lóbulo primario se relacionó de forma independiente con la enfermedad bilateral (oportunidad relativa, 7,61; intervalo de confianza 95 %, 2,44–23,8; P< 0,001). Entre los pacientes sin compromiso ganglionar desde el punto de vista clínico que presentaban carcinoma papilar sin multifocalidad tumoral en el lóbulo primario, 5 de 32 pacientes (16 %) tenían enfermedad bilateral. Los autores concluyeron que, en los niños con cáncer de tiroides diferenciado, la multifocalidad tumoral en el lóbulo primario se relaciona con enfermedad bilateral, y recomendaron considerar con rapidez una tiroidectomía completa después de la lobectomía inicial.
En otro análisis retrospectivo multicéntrico se evaluó la prevalencia y los factores de riesgo de la enfermedad multifocal en 212 pacientes pediátricos con carcinoma de tiroides papilar. La media de edad en el momento del diagnóstico fue de 14,1 años y 23 pacientes tenían 10 años o menos. Un total de 173 pacientes (82 %) eran mujeres. En 98 casos (46 %) existía enfermedad multifocal de cualquier grado, y en 73 casos (34 %) había enfermedad multifocal bilateral. Los factores predictivos de la enfermedad multifocal y multifocal bilateral fueron la edad de 10 años o menos, el estadio tumoral T3 y el estadio ganglionar N1b. Los autores llegaron a la conclusión de que deben tenerse en cuenta estos factores de riesgo y la alta prevalencia de la enfermedad multifocal al evaluar los riesgos y beneficios de las opciones de tratamiento quirúrgico en pacientes pediátricos con carcinoma de tiroides papilar.
La tiroidectomía total también permite mejorar el uso del yodo radiactivo para las pruebas de imágenes y el tratamiento.
- Disección central del cuello:
- Se debe realizar una disección central terapéutica de los ganglios linfáticos cuando hay indicios clínicos de metástasis centrales o laterales en el cuello.
- Se debe considerar una disección profiláctica central del cuello para pacientes sin indicios clínicos de invasión extratiroidea macroscópica o metástasis locorregional de acuerdo con la focalidad del tumor y el tamaño del tumor primario. Sin embargo, debido al aumento de mortalidad relacionada con la disección central de ganglios linfáticos, es importante evaluar caso por caso los riesgos y beneficios según el grado de la disección.
- Disección lateral del cuello:
- Se recomienda la confirmación citológica de enfermedad metastásica en los ganglios linfáticos laterales del cuello antes de la cirugía.
- No se recomienda la disección profiláctica rutinaria lateral de los ganglios linfáticos del cuello.
- Tiroidectomía:
- Clasificación y asignación del riesgo.
Pese a los datos probatorios limitados para el entorno pediátrico, la ATA Task Force recomienda el uso del sistema de clasificación tumor-ganglio-metástasis (TNM) para asignar a los pacientes a 1 de 3 grupos de riesgo. Esta estrategia de clasificación permite definir el riesgo de enfermedad cervical persistente y ayuda a determinar cuáles pacientes se deben someter a estadificación posoperatoria para establecer la presencia de metástasis a distancia.
- Riesgo pediátrico bajo según la ATA: enfermedad confinada a la tiroides con enfermedad N0 o NX, o pacientes con N1a diagnosticada de manera fortuita (metástasis microscópica en un número pequeño de ganglios centrales del cuello). Si bien estos pacientes tienen el riesgo más bajo de metástasis a distancia, aún tienen riesgo de enfermedad cervical residual; en particular, si la cirugía inicial no incluyó una disección central del cuello.
- Riesgo pediátrico intermedio según la ATA: enfermedad extensa N1a o enfermedad N1b mínima. Si bien estos pacientes tienen un riesgo bajo de metástasis a distancia, tienen un aumento del riesgo de una resección incompleta de ganglios linfáticos y de enfermedad cervical persistente.
- Riesgo pediátrico alto según la ATA: enfermedad regional extensa (N1b) o enfermedad localmente invasora (T4), con metástasis a distancia o sin esta. Los pacientes de este grupo tienen el riesgo más alto de resección incompleta, enfermedad persistente y metástasis a distancia.
Para obtener más información sobre el sistema TNM, consultar la sección Información sobre los estadios del cáncer de tiroides en Tratamiento del cáncer de tiroides.
- Estadificación posoperatoria y vigilancia a largo plazo.
La estadificación inicial se deberá realizar dentro de las 12 semanas posteriores a la cirugía para evaluar si hay indicios de enfermedad locorregional persistente e identificar a los pacientes con más probabilidades de beneficiarse de la terapia adicional con yodo I 131 (131I). Los niveles de riesgo pediátrico establecidos por la ATA (definidos antes) ayudan a determinar el alcance de las pruebas posoperatorias.
- Riesgo pediátrico bajo según la ATA:
- La estadificación posoperatoria inicial incluye una evaluación de la tiroglobulina con inhibición de la TSH. No se necesita una gammagrafía diagnóstica con yodo I 123 (123I).
- Se debe procurar obtener concentraciones de 0,5 a 1,0 mUI/l con la inhibición de la TSH.
- Para los pacientes sin indicios de enfermedad, la vigilancia debe incluir una ecografía a los 6 meses de la cirugía y, luego, cada año durante 5 años, así como una medición de las concentraciones de tiroglobulina (durante la terapia de reemplazo hormonal) cada 3 a 6 meses durante 2 años y, luego, cada año.
- Riesgo pediátrico intermedio según la ATA:
- La estadificación posoperatoria inicial incluye evaluación de la tiroglobulina con estimulación de la TSH y una gammagrafía diagnóstica de todo el cuerpo con 123I para estratificación y determinación con 131I posteriores.
- Se debe procurar obtener concentraciones de 0,1 a 0,5 mUI/l con la inhibición de la TSH.
- Para los pacientes sin indicios de enfermedad, la vigilancia debe incluir una ecografía a los 6 meses de la cirugía y, luego, cada 6 a 12 meses durante 5 años (después, con menor frecuencia), así como una medición de las concentraciones de tiroglobulina (durante la terapia de reemplazo hormonal) cada 3 a 6 meses durante 3 años y, luego, cada año.
- Se debe considerar la evaluación de la tiroglobulina con estimulación de la TSH y la gammagrafía diagnóstica con 123I al cabo de 1 o 2 años en los pacientes tratados con 131I.
- Riesgo pediátrico alto según la ATA:
- La estadificación posoperatoria inicial incluye evaluación de la tiroglobulina con estimulación de la TSH y una gammagrafía diagnóstica de todo el cuerpo con 123I para estratificación y determinación con 131I posteriores.
- Se debe procurar obtener concentraciones de 0,1 mUI/l con la inhibición de la TSH.
- Para los pacientes sin indicios de enfermedad, la vigilancia debe incluir una ecografía a los 6 meses de la cirugía y, luego, cada 6 a 12 meses durante 5 años (después, con menor frecuencia), así como una medición de las concentraciones de tiroglobulina (durante la terapia de remplazo hormonal) cada 3 a 6 meses durante 3 años y, luego, cada año.
- Se debe considerar la evaluación de la tiroglobulina con estimulación de la TSH y, posiblemente, una gammagrafía diagnóstica con 123I al cabo de 1 o 2 años en los pacientes tratados con 131I.
Para los pacientes con anticuerpos contra la tiroglobulina, se debe sopesar demorar la estadificación posoperatoria para dar tiempo a la depuración de anticuerpos, excepto para pacientes con enfermedad T4 o M1.
- Riesgo pediátrico bajo según la ATA:
- Ablación con yodo radiactivo.
La meta del tratamiento con 131I es disminuir la recidiva y la mortalidad mediante la eliminación de la enfermedad ávida de yodo.
- La ATA Task Force recomienda el uso de 131I para tratar la enfermedad locorregional ávida de yodo que es persistente o la enfermedad ganglionar que no se puede resecar, así como las metástasis distantes que se sabe o presume que están ávidas de yodo. Para los pacientes con enfermedad persistente después de la administración de 131I, se debe individualizar la decisión de continuar el tratamiento con 131I de acuerdo con los datos clínicos y la respuesta previa.
- Para facilitar la absorción de 131I por la enfermedad residual ávida de yodo, la concentración de la TSH debe ser superior a 30 mUI/l. Esta concentración se alcanza después de interrumpir la levotiroxina durante 14 días como mínimo. Para pacientes que no logran una respuesta adecuada de la TSH o no toleran un hipotiroidismo grave, se puede usar TSH humana recombinante.
- La administración terapéutica de 131I a menudo se basa en una dosificación empírica o una dosimetría de todo el cuerpo. La ATA Task Force no logró recomendar un abordaje específico porque no hay datos de comparación del tratamiento empírico con el tratamiento guiado por dosimetría. Sin embargo, debido a las diferencias de tamaño corporal y la depuración del yodo en los niños en comparación con los adultos, todas las actividades que involucren el uso de 131I deberían ser calculadas por personas expertas en la dosificación para niños.
- Para todos los niños, se recomienda una gammagrafía de todo el cuerpo 4 a 7 días después del tratamiento con 131I. La adición de una TC por emisión de fotón único con una TC convencional integrada (TCEFU-TC) puede ayudar a distinguir la ubicación anatómica de la absorción focal.
Los efectos tardíos del tratamiento con 131I, que son poco frecuentes, incluyen disfunción de las glándulas salivales, inhibición de la médula ósea, fibrosis pulmonar y segundas neoplasias malignas.
- En un estudio multicéntrico de niños y adolescentes con carcinoma de tiroides diferenciado, 285 pacientes consecutivos se trataron con tiroidectomía total y ablación con yodo radiactivo según las directrices de la ATA.
- El 87 % de los pacientes no tenía indicios de enfermedad activa al cabo de una mediana de seguimiento de 133 meses.
References
- Francis GL, Waguespack SG, Bauer AJ, et al.: Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 25 (7): 716-59, 2015.
- Spinelli C, Strambi S, Rossi L, et al.: Surgical management of papillary thyroid carcinoma in childhood and adolescence: an Italian multicenter study on 250 patients. J Endocrinol Invest 39 (9): 1055-9, 2016.
- Cherella CE, Richman DM, Liu E, et al.: Predictors of Bilateral Disease in Pediatric Differentiated Thyroid Cancer. J Clin Endocrinol Metab 106 (10): e4242-e4250, 2021.
- Banik GL, Shindo ML, Kraimer KL, et al.: Prevalence and Risk Factors for Multifocality in Pediatric Thyroid Cancer. JAMA Otolaryngol Head Neck Surg 147 (12): 1100-1106, 2021.
- Kim J, Sun Z, Adam MA, et al.: Predictors of nodal metastasis in pediatric differentiated thyroid cancer. J Pediatr Surg 52 (1): 120-123, 2017.
- Machens A, Elwerr M, Thanh PN, et al.: Impact of central node dissection on postoperative morbidity in pediatric patients with suspected or proven thyroid cancer. Surgery 160 (2): 484-92, 2016.
- Albano D, Bertagna F, Panarotto MB, et al.: Early and late adverse effects of radioiodine for pediatric differentiated thyroid cancer. Pediatr Blood Cancer 64 (11): , 2017.
- Cistaro A, Quartuccio N, Garganese MC, et al.: Prognostic factors in children and adolescents with differentiated thyroid carcinoma treated with total thyroidectomy and RAI: a real-life multicentric study. Eur J Nucl Med Mol Imaging 49 (4): 1374-1385, 2022.
Tratamiento del carcinoma de tiroides papilar y folicular recidivantes
A pesar de tener una enfermedad más avanzada en el momento de la presentación del cuadro clínico inicial que los adultos, los niños con cáncer de tiroides diferenciado tienen una supervivencia excelente y relativamente pocos efectos secundarios.
Las opciones de tratamiento para el carcinoma de tiroides papilar y folicular incluyen las siguientes:
- Ablación con yodo radiactivo 131 I (I 131).
Por lo general, la ablación con yodo radiactivo 131 I es efectiva después de una recidiva. Las terapias moleculares dirigidas que utilizan inhibidores de cinasas, pueden ser terapias alternativas en el caso de los pacientes con enfermedad resistente al tratamiento con 131 I.
Los inhibidores de tirosina–cinasas (ITC) con eficacia documentada para el tratamiento de adultos son los siguientes:
- Sorafenib. El sorafenib es un inhibidor del receptor del factor de crecimiento endotelial vascular (VEGFR), el receptor del factor de crecimiento derivado de plaquetas (PDGFR) y de la cinasa RAS. En un ensayo aleatorizado de fase III, el sorafenib mejoró la supervivencia sin progresión (SSP) en comparación con un placebo (10,8 vs. 5,8 meses) en pacientes adultos con cáncer de tiroides diferenciado resistente al yodo radiactivo y localmente avanzado o metastásico. La Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) aprobó el sorafenib en noviembre de 2013 para el tratamiento de adultos con carcinoma de tiroides diferenciado metastásico en estadio tardío.
Son limitados los datos específicos para pediatría; sin embargo, en un informe de caso, el sorafenib produjo una respuesta radiográfica en un paciente de 8 años con carcinoma de tiroides papilar metastásico.
- Lenvatinib. El lenvatinib es un inhibidor del VEGFR, el receptor del factor de crecimiento de fibroblastos, el PDGFR, así como de RET y KIT. En un estudio aleatorizado de fase III de adultos con cáncer de tiroides diferenciado resistente al tratamiento con I 131, el lenvatinib se relacionó con una mejora significativa de la SSP y la tasa de respuesta en comparación con un placebo. La FDA aprobó el lenvatinib en febrero de 2015 para el tratamiento de adultos con carcinoma de tiroides diferenciado progresivo resistente al yodo radiactivo.
Se notificó que 3 niños con carcinoma de tiroides papilar resistente al yodo radiactivo presentaron una respuesta clínica al lenvatinib.
- Inhibidores de BRAF. Se llevó a cabo un estudio sin anonimato, no aleatorizado de fase II sobre vemurafenib en adultos con carcinoma de tiroides que tenían una enfermedad resistente a 131I, metastásica o irresecable y positiva para la mutación BRAFV600E. Ningún paciente había recibido tratamiento con un TCI. Se documentó una tasa de respuesta del 38,5 %. La combinación de dabrafenib con el inhibidor de MEK, trametinib, mostró una tasa de respuesta del 69 % en los pacientes con carcinoma de tiroides anaplásico metastásico o avanzado con la mutación BRAF V600E.
- Larotrectinib y entrectinib (inhibidores de NTRK). El larotrectinib (terapia dirigida) se ha usado para el tratamiento de pacientes con carcinoma de tiroides positivo para la fusión de TRK. Los 5 pacientes con carcinomas de tiroides positivos para la fusión de TRK que recibieron larotrectinib exhibieron respuestas parciales o completas. También se han notificado respuestas con el entrectinib. La FDA aprobó el larotrectinib y el entrectinib para el tratamiento de adultos y niños (entrectinib está limitado a los pacientes mayores de 12 años) con tumores sólidos que tengan todas las siguientes características:
- Tienen un gen de fusión NTRK sin una mutación con resistencia adquirida conocida.
- Son metastásicos o son tumores para los que una resección quirúrgica probablemente resultará en morbilidad grave.
- Carecen de tratamientos alternativos satisfactorios o han progresado después del tratamiento.
- Selpercatinib (inhibidor de RET). En un ensayo de fase I/II de terapia con selpercatinib para pacientes (intervalo de edad, 25–88 años) con cánceres con mutación en RET, se inscribió a 19 pacientes con fusión de RET tratados antes por cánceres de tiroides.
- De los 19 pacientes, 15 (79 %) alcanzaron una respuesta objetiva (1 respuesta completa y 14 respuestas parciales) y la mediana de duración de la respuesta fue de 18,4 meses.
- Los efectos adversos de grados 3 a 4 relacionados con el tratamiento fueron hipertensión (12 %), aumento de la alanina–aminotransferasa (ALT) (10 %) y aspartato–aminotransferasa (AST) (7 %), diarrea (3 %) y prolongación del intervalo QT (2 %).
- La FDA aprobó el selpercatinib (mediante aprobación acelerada) para el tratamiento de pacientes adultos y pediátricos de 12 años y más con cáncer de tiroides con fusión de RET en estadio avanzado o metastático para los que es necesario terapia sistémica y son resistentes al yodo radiactivo (si el yodo radiactivo fuera apropiado).
- Cabozantinib (inhibidor de VEGFR y RET). En un ensayo aleatorizado con enmascaramiento doble de fase III (COSMIC-311 [NCT03690388]) de pacientes adultos (intervalo de edad, 55–72 años) se comparó el cabozantinib con un placebo. Los pacientes habían recibido por lo menos una tirosina cinasa dirigida a VEGFR para el carcinoma de tiroides diferenciado, y su enfermedad se consideró progresiva y resistente al yodo radioactivo. Se observó una respuesta eficaz en 10 de 67 pacientes que recibieron cabozantinib, en comparación con cero respuestas en el grupo de placebo. La SSP fue de 11 meses (intervalo de confianza [IC] 95 %, 7,4–13,8) en el grupo de cabozantinib, en comparación con 1,9 meses (IC 95 %, 1,9–3,7) en el grupo de placebo, con un cociente de riesgos instantáneos de 0,22 (IC 95 %, 0,14–0,31). Con base en estos datos, la FDA aprobó el cabozantinib en el tratamiento de esta población.
Para obtener más información, consultar Tratamiento del cáncer de tiroides.
Opciones de tratamiento en evaluación clínica del carcinoma de tiroides papilar y folicular recidivante
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de ensayo clínico nacional o institucional en curso:
- LIBRETTO-121 (NCT03899792) (A Study of Oral LOXO-292 [Selpercatinib] in Pediatric Participants With Advanced Solid or Primary Central Nervous System [CNS] Tumors): este estudio multicéntrico sin anonimato de fase I/II de LOXO-292 oral en pacientes pediátricos con una alteración activadora en RET y un tumor sólido avanzado o un tumor primario del SNC.
References
- Golpanian S, Perez EA, Tashiro J, et al.: Pediatric papillary thyroid carcinoma: outcomes and survival predictors in 2504 surgical patients. Pediatr Surg Int 32 (3): 201-8, 2016.
- Vergamini LB, Frazier AL, Abrantes FL, et al.: Increase in the incidence of differentiated thyroid carcinoma in children, adolescents, and young adults: a population-based study. J Pediatr 164 (6): 1481-5, 2014.
- Dermody S, Walls A, Harley EH: Pediatric thyroid cancer: An update from the SEER database 2007-2012. Int J Pediatr Otorhinolaryngol 89: 121-6, 2016.
- Powers PA, Dinauer CA, Tuttle RM, et al.: Treatment of recurrent papillary thyroid carcinoma in children and adolescents. J Pediatr Endocrinol Metab 16 (7): 1033-40, 2003.
- Brose MS, Nutting CM, Jarzab B, et al.: Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet 384 (9940): 319-28, 2014.
- Iyer P, Mayer JL, Ewig JM: Response to sorafenib in a pediatric patient with papillary thyroid carcinoma with diffuse nodular pulmonary disease requiring mechanical ventilation. Thyroid 24 (1): 169-74, 2014.
- Schlumberger M, Tahara M, Wirth LJ, et al.: Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med 372 (7): 621-30, 2015.
- Mahajan P, Dawrant J, Kheradpour A, et al.: Response to Lenvatinib in Children with Papillary Thyroid Carcinoma. Thyroid 28 (11): 1450-1454, 2018.
- Brose MS, Cabanillas ME, Cohen EE, et al.: Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol 17 (9): 1272-82, 2016.
- Subbiah V, Kreitman RJ, Wainberg ZA, et al.: Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. J Clin Oncol 36 (1): 7-13, 2018.
- Drilon A, Laetsch TW, Kummar S, et al.: Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med 378 (8): 731-739, 2018.
- Chu YH, Dias-Santagata D, Farahani AA, et al.: Clinicopathologic and molecular characterization of NTRK-rearranged thyroid carcinoma (NRTC). Mod Pathol 33 (11): 2186-2197, 2020.
- Bayer HealthCare Pharmaceuticals: VITRAKVI (larotrectinib): Prescribing Information. Stamford, Conn: Loxo Oncology, Inc., 2018. Available online. Last accessed September 28, 2020.
- Wirth LJ, Sherman E, Robinson B, et al.: Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N Engl J Med 383 (9): 825-835, 2020.
- Eli Lilly and Company: RETEVMO (selpercatinib): Prescribing Information. Indianapolis, Ind: Lilly USA, LLC, 2020. Available online. Last accessed September 28, 2020.
- Brose MS, Robinson B, Sherman SI, et al.: Cabozantinib for radioiodine-refractory differentiated thyroid cancer (COSMIC-311): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 22 (8): 1126-1138, 2021.
- Duke ES, Barone AK, Chatterjee S, et al.: FDA Approval Summary: Cabozantinib for Differentiated Thyroid Cancer. Clin Cancer Res 28 (19): 4173-4177, 2022.
Tratamiento del carcinoma de tiroides medular
Los carcinomas de tiroides medular por lo general se relacionan con el síndrome de neoplasia endocrina múltiple de tipo 2 (NEM2). Para obtener más información, consultar Tratamiento de los síndromes de neoplasia endocrina múltiple infantiles.
Las opciones de tratamiento del carcinoma de tiroides medular son las siguientes:
- Cirugía. El tratamiento de los niños con carcinoma de tiroides medular es sobre todo quirúrgico. Los investigadores concluyeron que la disección profiláctica de los ganglios linfáticos centrales no se debería realizar en pacientes de cáncer de tiroides medular hereditario si sus concentraciones séricas basales de calcitonina son inferiores a 40 pg/ml.
La mayoría de los casos de carcinoma de tiroides medular en niños se presentan en el contexto de los síndromes NEM2A y NEM2B. En esos casos familiares, se indican pruebas y asesoramiento genético tempranos, y se recomienda cirugía profiláctica para los niños con una mutación de la línea germinal en RET. Las fuertes correlaciones entre genotipo y fenotipo facilitaron la formulación de directrices de intervención, incluso sobre los exámenes de detección y la edad en que se debería realizar la tiroidectomía profiláctica.
En un análisis retrospectivo se identificaron 167 niños con mutaciones en el gen RET que se sometieron a tiroidectomía profiláctica. Este grupo incluyó a 109 pacientes sin disección simultánea de los ganglios linfáticos centrales y 58 pacientes con disección simultánea de los ganglios linfáticos centrales. El hipoparatiroidismo posoperatorio fue más frecuente en niños mayores (32 % en el grupo de mayor edad vs. 3 % en el grupo de menor edad; P = 0,002), independientemente de si se realizó la disección de los ganglios linfáticos centrales. Tres niños desarrollaron parálisis del nervio laríngeo recurrente, todos se sometieron a disección de los ganglios linfáticos centrales (P = 0,040). Todas las complicaciones se solucionaron en 6 meses. La normalización de las concentraciones de calcitonina sérica posoperatoria fue posible en 114 de los 115 niños (99,1 %) que presentaron un aumento en los valores preoperatorios. Los niños se clasificaron en grupos de riesgo según el tipo específico de mutación en el gen RET (consultar el Cuadro 1).
- En la categoría de riesgo más alto, se encontró carcinoma de tiroides medular en 5 de 6 niños (83 %) de 3 años o menores.
- En la categoría de riesgo alto, se encontró carcinoma de tiroides medular en 6 de 20 niños (30 %) de 3 años o menores, en 16 de 36 niños (44 %) de 4 a 6 años, y en 11 de 16 niños (69 %) de 7 a 12 años (P = 0,081).
- En la categoría de riesgo moderado, se observó carcinoma de tiroides medular en 1 de 9 niños (11 %) de 3 años o menores, en 1 de 26 niños (4 %) de 4 a 6 años, en 3 de 26 niños (12 %) de 7 a 12 años, y en 7 de 16 niños (44 %) de 13 a 18 años (P = 0,006).
La American Thyroid Association propuso las siguientes directrices para la tiroidectomía profiláctica en niños con carcinoma de tiroides medular hereditario (consultar el Cuadro 1).
Cuadro 1. Categorías de riesgo y tratamiento a partir de mutaciones comunes en RET detectadas en el examen genéticoa
Categoría de riesgo de carcinoma de tiroides medular Más alto (NEM2B) Alto (NEM2A) Moderado (NEM2A) NEM2A = neoplasia endocrina múltiple de tipo 2A; NEM2B = neoplasia endocrina múltiple de tipo 2B. aAdaptado de Wells et al. Mutación en RET M918T A883F, C634F/G/R/S/W/Y G533C, C609F/G/R/S/Y, C611F/G/S/Y/W, C618F/R/S, C620F/R/S, C630R/Y, D631Y, K666E, E768D, L790F, V804L, V804M, S891A, R912P Edad para la tiroidectomía profiláctica Tiroidectomía total en el primer año de vida, idealmente en los primeros meses de vida. Tiroidectomía total a los 5 años de edad o antes según las concentraciones de calcitonina sérica. Tiroidectomía total cuando las concentraciones de calcitonina sérica excedan los límites normales o en otro momento oportuno si los padres prefieren evitar un período de vigilancia largo. - Terapia con inhibidor de tirosina–cinasas (ITC). Se evaluaron y aprobaron varios ITC para pacientes con cáncer de tiroides medular en estadio avanzado.
- Vandetanib. La Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) aprobó el vandetanib (un inhibidor de la cinasa de RET, del receptor del factor de crecimiento endotelial vascular [VEGFR] y de la señalización del receptor del factor de crecimiento epidérmico) para el tratamiento del cáncer de tiroides medular sintomático o progresivo en pacientes adultos con enfermedad irresecable, localmente avanzada o metastásica. Su aprobación se fundamentó en un ensayo de fase III aleatorizado y controlado con placebo, en el que se observó una mejoría notable de la supervivencia sin progresión (SSP) en los pacientes asignados al azar para recibir vandetanib (cociente de riesgos instantáneos, 0,35). En el ensayo también se observó una ventaja de la tasa de respuesta objetiva para los pacientes que recibieron vandetanib (44 vs. 1 % para el grupo de placebo).
En un ensayo de fase I/II, se trataron con vandetanib a los niños con carcinoma de tiroides medular metastásico localmente avanzado o con carcinoma de tiroides medular metastásico. De 16 pacientes, solo 1 no respondió al tratamiento y 7 presentaron una respuesta parcial con una tasa de respuesta objetiva del 44 %. La enfermedad recidivó en 3 de esos pacientes, pero 11 de 16 pacientes tratados con vandetanib seguían recibiendo la terapia en el momento del informe. La duración de la terapia en la cohorte completa osciló entre 2 y 52 meses, con una mediana de 27 meses. Una evaluación a largo plazo de una cohorte de 17 niños y adolescentes con carcinoma de tiroides medular avanzado que recibieron vandetanib, informó una media de SSP de 6,7 años y una supervivencia general a 5 años del 88,2 %.
- Cabozantinib. El cabozantinib (un inhibidor de las cinasas RET y MET y del VEGFR) también mostró actividad contra el cáncer de tiroides medular irresecable (10 a 35 pacientes adultos [29 %] presentaron una respuesta parcial). La FDA aprobó el cabozantinib en 2012 para el tratamiento de adultos con cáncer de tiroides medular metastásico.
- Selpercatinib. En un ensayo de fase I/II de terapia con selpercatinib (inhibidor de RET) para pacientes con cánceres con la mutación en RET, se inscribió a 55 pacientes de cáncer de tiroides medular con mutación en RET (intervalo de edad, 17–84 años) que se habían tratado antes con vandetanib o cabozantinib y a 88 pacientes de cáncer de tiroides medular con mutación en RET (intervalo de edad, 15–82 años) que no se habían tratado antes con vandetanib o cabozantinib.
- En la cohorte que se había tratado antes, el 69 % de los pacientes alcanzaron una respuesta objetiva y, tras una mediana de seguimiento de 14 meses, la mediana de duración de la respuesta no se había alcanzado.
- En la cohorte de pacientes sin tratamiento previo, el 73 % de los pacientes alcanzaron una respuesta objetiva con una mediana de duración de la respuesta de 22 meses.
- Los efectos adversos de grados 3 o 4 más comunes fueron hipertensión (12 %), aumento de la alanina– aminotransferasa (ALT) (10 %) y la aspartato–aminotransferasa (AST) (7 %), diarrea (3 %) y prolongación del intervalo QT (2 %).
- En una cohorte pequeña de 6 niños con carcinoma de tiroides medular recidivante tratados con selpercatinib, todos los pacientes presentaron respuestas duraderas al cabo de una mediana de seguimiento de 13 meses.
- La FDA aprobó el selpercatinib (mediante aprobación acelerada) para el tratamiento de pacientes adultos y pediátricos de 12 años y más con cáncer de tiroides medular en estadio avanzado o metastásico con mutación en RET que necesiten terapia sistémica.
- Vandetanib. La Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) aprobó el vandetanib (un inhibidor de la cinasa de RET, del receptor del factor de crecimiento endotelial vascular [VEGFR] y de la señalización del receptor del factor de crecimiento epidérmico) para el tratamiento del cáncer de tiroides medular sintomático o progresivo en pacientes adultos con enfermedad irresecable, localmente avanzada o metastásica. Su aprobación se fundamentó en un ensayo de fase III aleatorizado y controlado con placebo, en el que se observó una mejoría notable de la supervivencia sin progresión (SSP) en los pacientes asignados al azar para recibir vandetanib (cociente de riesgos instantáneos, 0,35). En el ensayo también se observó una ventaja de la tasa de respuesta objetiva para los pacientes que recibieron vandetanib (44 vs. 1 % para el grupo de placebo).
Para obtener más información, consultar Tratamiento de los síndromes de neoplasia endocrina múltiple infantiles y la sección en inglés Treatment for Medullary Thyroid Cancer [MTC] en Genetics of Endocrine and Neuroendocrine Neoplasias.
Opciones de tratamiento en evaluación clínica para el carcinoma de tiroides medular
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de ensayo clínico nacional o institucional en curso:
- LIBRETTO-121 (NCT03899792) (A Study of Oral LOXO-292 [Selpercatinib] in Pediatric Participants With Advanced Solid or Primary Central Nervous System [CNS] Tumors): este estudio multicéntrico sin anonimato de fase I/II de LOXO-292 oral en pacientes pediátricos con una alteración activadora en RET y un tumor sólido avanzado o un tumor primario del SNC.
References
- Machens A, Elwerr M, Thanh PN, et al.: Impact of central node dissection on postoperative morbidity in pediatric patients with suspected or proven thyroid cancer. Surgery 160 (2): 484-92, 2016.
- Wells SA, Asa SL, Dralle H, et al.: Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 25 (6): 567-610, 2015.
- Machens A, Elwerr M, Lorenz K, et al.: Long-term outcome of prophylactic thyroidectomy in children carrying RET germline mutations. Br J Surg 105 (2): e150-e157, 2018.
- Wells SA, Robinson BG, Gagel RF, et al.: Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol 30 (2): 134-41, 2012.
- Thornton K, Kim G, Maher VE, et al.: Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: U.S. Food and Drug Administration drug approval summary. Clin Cancer Res 18 (14): 3722-30, 2012.
- Fox E, Widemann BC, Chuk MK, et al.: Vandetanib in children and adolescents with multiple endocrine neoplasia type 2B associated medullary thyroid carcinoma. Clin Cancer Res 19 (15): 4239-48, 2013.
- Kraft IL, Akshintala S, Zhu Y, et al.: Outcomes of Children and Adolescents with Advanced Hereditary Medullary Thyroid Carcinoma Treated with Vandetanib. Clin Cancer Res 24 (4): 753-765, 2018.
- Kurzrock R, Sherman SI, Ball DW, et al.: Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol 29 (19): 2660-6, 2011.
- Wirth LJ, Sherman E, Robinson B, et al.: Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N Engl J Med 383 (9): 825-835, 2020.
- Shankar A, Kurzawinski T, Ross E, et al.: Treatment outcome with a selective RET tyrosine kinase inhibitor selpercatinib in children with multiple endocrine neoplasia type 2 and advanced medullary thyroid carcinoma. Eur J Cancer 158: 38-46, 2021.
- Eli Lilly and Company: RETEVMO (selpercatinib): Prescribing Information. Indianapolis, Ind: Lilly USA, LLC, 2020. Available online. Last accessed September 28, 2020.
Actualizaciones más recientes a este resumen (1/23/2024)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Se incorporaron cambios editoriales en este resumen.
El Consejo editorial del PDQ sobre el tratamiento pediátrico es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este resumen del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento del cáncer de tiroides infantil. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El Consejo editorial del PDQ sobre el tratamiento pediátrico, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
- Si el artículo se debe analizar en una reunión del consejo.
- Si conviene añadir texto acerca del artículo.
- Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Tratamiento del cáncer de tiroides infantil son:
- Denise Adams, MD (Children's Hospital Boston)
- Karen J. Marcus, MD, FACR (Dana-Farber Cancer Institute/Boston Children's Hospital)
- William H. Meyer, MD
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Alberto S. Pappo, MD (St. Jude Children's Research Hospital)
- D. Williams Parsons, MD, PhD (Texas Children's Hospital)
- Arthur Kim Ritchey, MD (Children's Hospital of Pittsburgh of UPMC)
- Carlos Rodriguez-Galindo, MD (St. Jude Children's Research Hospital)
- Stephen J. Shochat, MD (St. Jude Children's Research Hospital)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El Consejo editorial del PDQ sobre el tratamiento pediátrico emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como “En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]”.
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
PDQ® sobre el tratamiento pediátrico. PDQ Tratamiento del cáncer de tiroides infantil. Bethesda, MD: National Cancer Institute. Actualización:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como “estándar” o “en evaluación clínica”. Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Comuníquese con el Instituto Nacional del Cáncer
Para obtener más información sobre las opciones para comunicarse con el NCI, incluso la dirección de correo electrónico, el número telefónico o el chat, consultar la página del Servicio de Información de Cáncer del Instituto Nacional del Cáncer.